Posts
My thoughts and ideas
Welcome to the blog
My thoughts and ideas
Introduction to bioinformatics for RNA sequence analysis
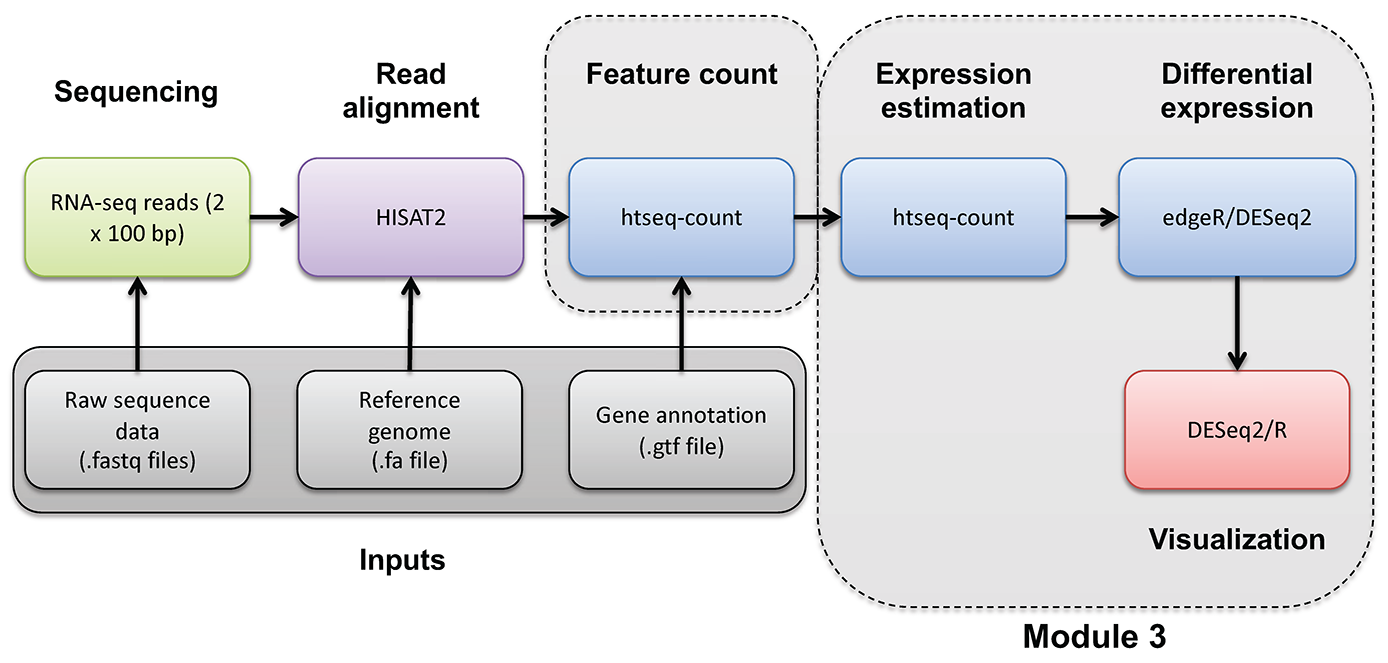
If you would like a brief refresher on differential expression analysis, please refer to the mini lecture.
In this tutorial you will:
First, create a directory for results. Then start R:
cd $RNA_HOME/
mkdir -p de/deseq2
cd de/deseq2
R
Once we start from R, the relevant packages should already be installed. We will load the libraries, set working directories, and read in the raw read counts data. Two pieces of information are required to perform analysis with DESeq2. A matrix of raw counts, such as was generated previously while running HTseq previously in this course. This is important as DESeq2 normalizes the data, correcting for differences in library size using using this type of data. DESeq2 also requires the experimental design which can be supplied as a data.frame, detailing the samples and conditions.
# define working dir paths
datadir = "/home/ubuntu/workspace/rnaseq/expression/htseq_counts"
outdir = "/home/ubuntu/workspace/rnaseq/de/deseq2"
# load R libraries we will use in this section
library(DESeq2)
library(data.table)
library(ggplot2)
# set working directory to data dir
setwd(datadir)
# read in the RNAseq read counts for each gene (produced by htseq-count)
htseqCounts = fread("gene_read_counts_table_all_final.tsv")
# set working directory to the output dir
setwd(outdir)
DESeq2 has a number of options for data import and has a function to read HTseq output files directly. Here the most universal option is used, reading in raw counts from a matrix in simple TSV format (one row per gene, one column per sample). The HTseq count data that was read in above is stored as an object of class “data.table”, this can be verified with the class() function. Before use in this exercise it is required to convert this object to an appropriate matrix format with gene names as rows and samples as columns.
It should be noted that while the replicate samples are technical replicates (i.e. the same library was sequenced), herein they are treated as biological replicates for illustrative purposes. DESeq2 does have a function to collapse technical replicates though.
# view the class of the data
class(htseqCounts)
# convert the data.table to matrix format
htseqCounts = as.matrix(htseqCounts)
class(htseqCounts)
# set the gene ID values to be the row names for the matrix
rownames(htseqCounts) = htseqCounts[, "GeneID"]
# now that the gene IDs are the row names, remove the redundant column that contains them
htseqCounts = htseqCounts[, colnames(htseqCounts) != "GeneID"]
# convert the count values from strings (with spaces) to integers, because originally the gene column contained characters, the entire matrix was set to character
class(htseqCounts) = "integer"
# view the first few lines of the gene count matrix
head(htseqCounts)
Before running DESeq2 (or any differential expression analysis) it is useful to pre-filter data. There are computational benefits to doing this as the memory size of the objects within R will decrease and DESeq2 will have less data to work through and will be faster. By removing “low quality” data, it is also reduces the number of statistical tests that will be performed, which in turn reduces the impact of multiple test correction and can lead to more significant genes.
The amount of pre-filtering is up to the analyst however, it should be done in an unbiased way. DESeq2 recommends removing any gene with less than 10 reads across all samples. Below, we filter a gene if at least 1 sample does not have at least 10 reads. Either way, mostly what is being removed here are genes with very little evidence for expression in any sample (in many cases gene with 0 counts in all samples).
# run a filtering step
# i.e. require that for every gene: at least 1 of 6 samples must have counts greater than 10
# get index of rows that meet this criterion and use that to subset the matrix
# note the dimensions of the matrix before and after filtering with dim()
# breaking apart the command below to understand it's outcome
tail(htseqCounts) # look at the raw counts
tail(htseqCounts >= 10) # determine which cells have counts greater than 10
tail(rowSums(htseqCounts >= 10)) # determine for which genes how many samples have counts greater than 10
tail(rowSums(htseqCounts >= 10) >= 1) # filter to those entries/genes for which at least one sample has counts greater than 10
tail(which(rowSums(htseqCounts >= 10) >= 1)) #obtain the index for the above filter criteria
dim(htseqCounts)
htseqCounts = htseqCounts[which(rowSums(htseqCounts >= 10) >= 1), ]
dim(htseqCounts)
As mentioned above DESeq2 also needs to know the experimental design, that is which samples belong to which condition to test. The experimental design for the example dataset herein is quite simple as there are 6 samples with two conditions to compare (UHR vs HBR), as such we can just create the experimental design right within R. There is one important thing to note, DESeq2 does not check sample names, it expects that the column names in the counts matrix we created correspond exactly to the row names we specify in the experimental design.
# construct a mapping of the meta data for our experiment (comparing UHR cell lines to HBR brain tissues)
# this is defining the biological condition/label for each experimental replicate
# create a simple one column dataframe to start
metaData <- data.frame("Condition" = c("UHR", "UHR", "UHR", "HBR", "HBR", "HBR"))
# convert the "Condition" column to a factor data type
# the arbitrary order of these factors will determine the direction of log2 fold-changes for the genes (i.e. up or down regulated)
metaData$Condition = factor(metaData$Condition, levels = c("HBR", "UHR"))
# set the row names of the metaData dataframe to be the names of our sample replicates from the read counts matrix
rownames(metaData) = colnames(htseqCounts)
# view the metadata dataframe
head(metaData)
# check that names of htseq count columns match the names of the meta data rows
# use the "all" function which tests whether an entire logical vector is TRUE
all(rownames(metaData) == colnames(htseqCounts))
With all the data properly formatted it is now possible to combine all the information required to run differental expression in one object. This object will hold the input data, and intermediary calculations. It is also where the condition to test is specified.
# make deseq2 data sets
# here we are setting up our experiment by supplying: (1) the gene counts matrix, (2) the sample/replicate for each column, and (3) the biological conditions we wish to compare.
# this is a simple example that works for many experiments but these can also get more complex
# for example, including designs with multiple variables such as "~ group + condition",
# and designs with interactions such as "~ genotype + treatment + genotype:treatment".
dds = DESeqDataSetFromMatrix(countData = htseqCounts, colData = metaData, design = ~Condition)
# the design formula above is often a point of confussion, it is useful to put in words what is happening, when we specify "design = ~Condition" we are saying
# regress gene expression on condition, or put another way model gene expression on condition
# gene expression is the response variable and condition is the explanatory variable
# you can put words to formulas using this [cheat sheet](https://www.econometrics.blog/post/the-r-formula-cheatsheet/)
With all the data now in place, DESeq2 can be run. Calling DESeq2 will perform the following actions:
?results and this paper for details)# run the DESeq2 analysis on the "dds" object
dds = DESeq(dds)
# view the first 5 lines of the DE results
res = results(dds)
head(res, 5)
It is good practice to shrink the log-fold change values, this does exactly what it sounds like, reducing the amount of log-fold change for genes where there are few counts which create huge variability that is not truly biological signal. Consider for example a gene for two samples, one sample has 1 read, and and one sample has 6 reads, that is a 6 fold change, that is likely not accurate. There are a number of algorithms that can be used to shrink log2 fold change, here we will use the apeglm algorithm, which does require the apeglm package to be installed.
# shrink the log2 fold change estimates
# shrinkage of effect size (log fold change estimates) is useful for visualization and ranking of genes
# In simplistic terms, the goal of calculating "dispersion estimates" and "shrinkage" is also to account for the problem that
# genes with low mean counts across replicates tend to have higher variability than those with higher mean counts.
# Shrinkage attempts to correct for this. For a more detailed discussion of shrinkage refer to the DESeq2 vignette
# first get the name of the coefficient (log fold change) to shrink
resultsNames(dds)
# now apply the shrinkage approach
resLFC = lfcShrink(dds, coef = "Condition_UHR_vs_HBR", type = "apeglm")
# make a copy of the shrinkage results to manipulate
deGeneResult = resLFC
# contrast the values before and after shrinkage
# create a temporary, simplified data structure with the values before/after shrinkage, and create a new column called "group" with this label
res_before = res
res_before$group = "before shrinkage"
res_after = deGeneResult
res_after$group = "after shrinkage"
res_combined = rbind(res_before[,c("log2FoldChange","group")], res_after[,c("log2FoldChange","group")])
# adjust the order so that the legend has "before shrinkage" listed first
res_combined$group = factor(res_combined$group, levels = c("before shrinkage", "after shrinkage"))
# look at the fold change values
head(res_before)
head(res_after)
p <- ggplot(res_combined, aes(x = log2FoldChange, color = group)) + geom_density() + theme_bw() +
scale_color_manual(values = c("before shrinkage" = "tomato4", "after shrinkage" = "slategray")) +
labs(color = "Shrinkage status")
ggsave(plot=p, filename = "before_after_shrinkage.pdf", device="pdf", width=6, height=4, units="in")
How did the results change before and after shinkage? What direction is each log2 fold change value moving?
Note that for these data, the impact of shrinkage is very subtle but the pattern is that fold change values move towards 0.
DESeq2 was run with ensembl gene IDs as identifiers, this is not the most human friendly way to interpret results. Here gene symbols are merged onto the differential expressed gene list to make the results a bit more interpretable.
# read in gene ID to name mappings (using "fread" an alternative to "read.table")
gene_id_mapping <- fread("/home/ubuntu/workspace/rnaseq/de/htseq_counts/ENSG_ID2Name.txt", header = FALSE)
# add names to the columns in the "gene_id_mapping" dataframe
setnames(gene_id_mapping, c("ensemblID", "Symbol"))
# view the first few lines of the gene ID to name mappings
head(gene_id_mapping)
# merge on gene names
deGeneResult$ensemblID = rownames(deGeneResult)
deGeneResult = as.data.table(deGeneResult)
deGeneResult = merge(deGeneResult, gene_id_mapping, by = "ensemblID", all.x = TRUE)
# merge the original raw count values onto this final dataframe to aid interpretation
original_counts = as.data.frame(htseqCounts)
original_counts[,"ensemblID"] = rownames(htseqCounts)
deGeneResult = merge(deGeneResult, original_counts, by = 'ensemblID', all.x = TRUE)
# view the final merged dataframe
# based on the raw counts and fold change values what does a negative fold change mean with respect to our two conditions HBR and UHR?
head(deGeneResult)
With the DE analysis complete it is useful to view and filter the data frames to only the relevant genes. Here some basic data manipulation is performed, filtering to significant genes at specific thresholds.
# view the top genes according to adjusted p-value
deGeneResult[order(deGeneResult$padj), ]
# view the top genes according to fold change
deGeneResult[order(deGeneResult$log2FoldChange), ]
# determine the number of up/down significant genes at FDR < 0.05 significance level
dim(deGeneResult)[1] # number of genes tested
dim(deGeneResult[deGeneResult$padj < 0.05])[1] #number of significant genes
# order the DE results by adjusted p-value
deGeneResultSorted = deGeneResult[order(deGeneResult$padj), ]
# create a filtered data frame that limits to only the significant DE genes (adjusted p.value < 0.05)
deGeneResultSignificant = deGeneResultSorted[deGeneResultSorted$padj < 0.05]
The data generated is now written out as tab separated files. Some of the DESeq2 objects are also saved as serialized R (RDS) objects which can be read back into R later for visualization.
# set the working directory to the output dir where we will store any results files
setwd(outdir)
# save the final DE result (all genes) to an output file
fwrite(deGeneResultSorted, file="DE_all_genes_DESeq2.tsv", sep="\t")
# save the final DE result (significant genes only) to an output file
fwrite(deGeneResultSignificant, file="DE_sig_genes_DESeq2.tsv", sep="\t")
# save the DESeq2 objects for the data visualization section
saveRDS(dds, "dds.rds")
saveRDS(res, "res.rds")
saveRDS(resLFC, "resLFC.rds")
For both conditions (HBR and UHR) lets take a look at the top n genes but this time according to fold-change instead of p-value.
# use the dplyr library to manipulate the dataframe
library(dplyr)
# create a new copy of the data frame, sorted by log2 fold change
deGeneResultSortedFoldchange = arrange(deGeneResultSignificant, log2FoldChange)
# create a convenient data structure with just the top n genes from each condition
top_bottom = bind_rows(
head(deGeneResultSortedFoldchange, 10) %>% mutate(Set = "Bottom 10"),
tail(deGeneResultSortedFoldchange, 10) %>% mutate(Set = "Top 10")
)
# visualize data for the top n genes. Simplify the output a bit
print(top_bottom[,c("log2FoldChange","padj","Symbol","UHR_Rep1","UHR_Rep2","UHR_Rep3","HBR_Rep1","HBR_Rep2","HBR_Rep3","Set")])
#To exit R type the following
quit(save = "no")
Download the file: outdir/DE_sig_genes_DESeq2.tsv. Open this spreadsheet, sort on “log2FoldChange” column and find the top 100 significant genes with higher expression in HBR (brain). Also download the file: outdir/DE_all_genes_DESeq2.tsv (to be used as a list of background genes or where we want the fold-change value for every gene).
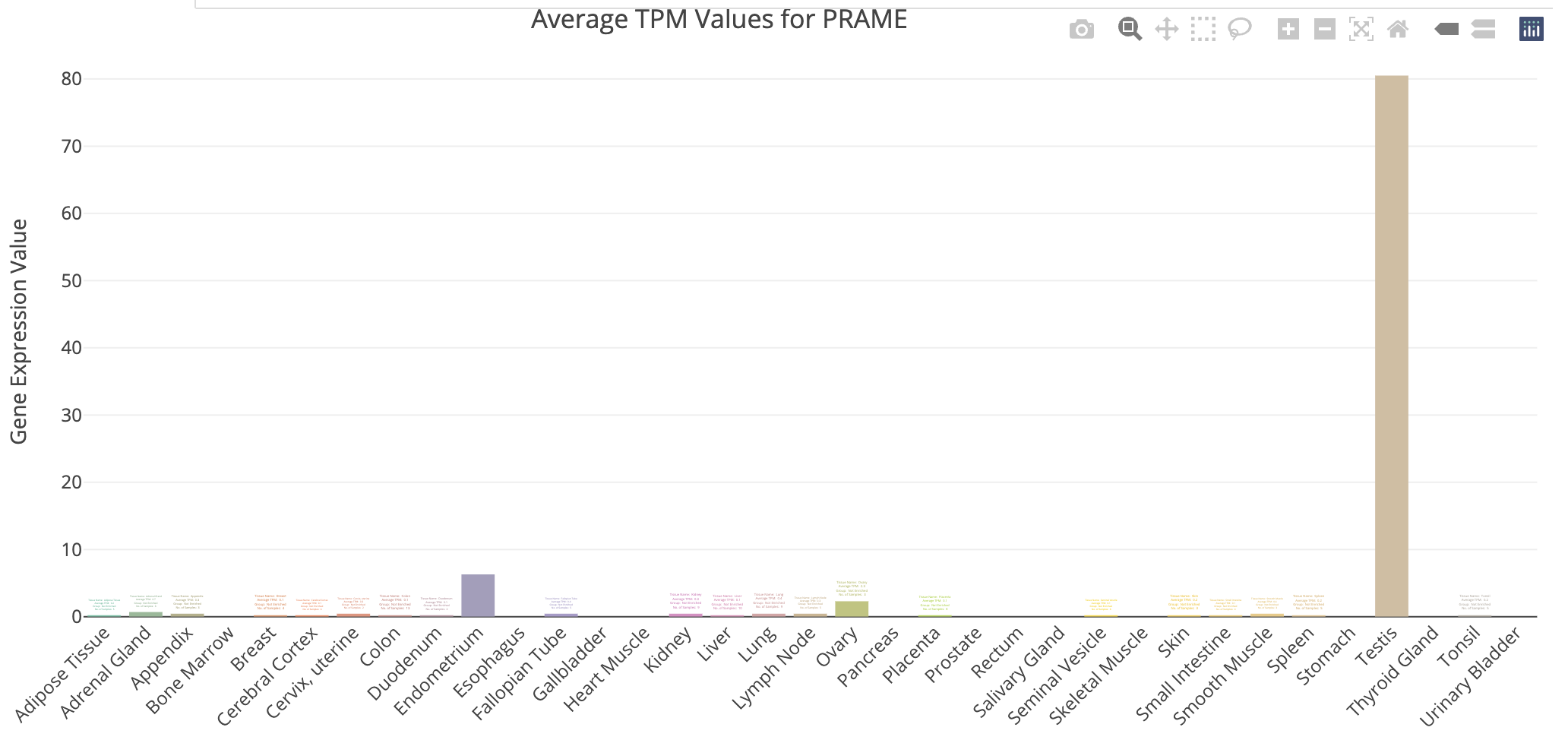
DE_all_genes_DESeq2.tsv for this purpose. You can also manually explore some individual genes over-expressed in UHR with the Tissue-Specific Genes tool. For example, try PRAME and SERPIND1, two of the top UHR genes.g:Profiler example result
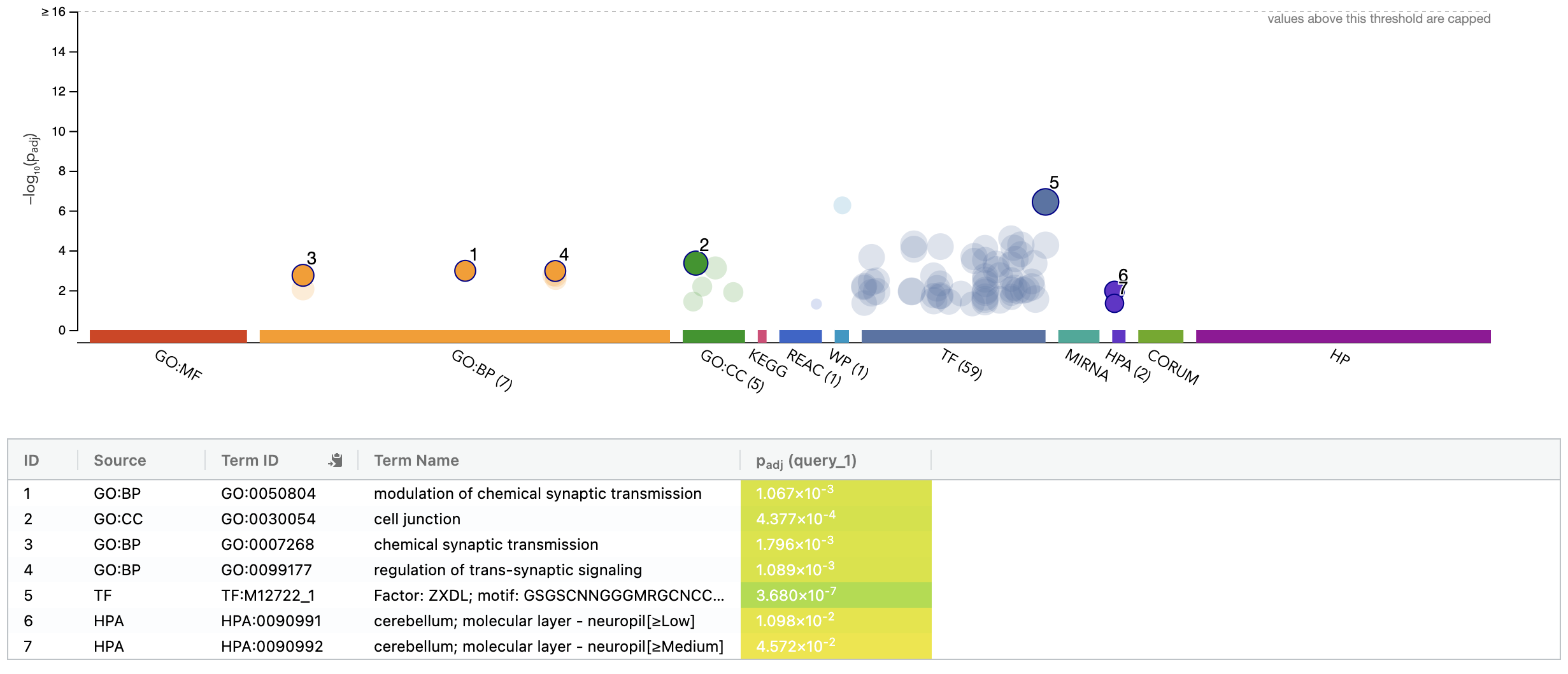
TissueEnrich summary for top 100 HBR over-expressed genes
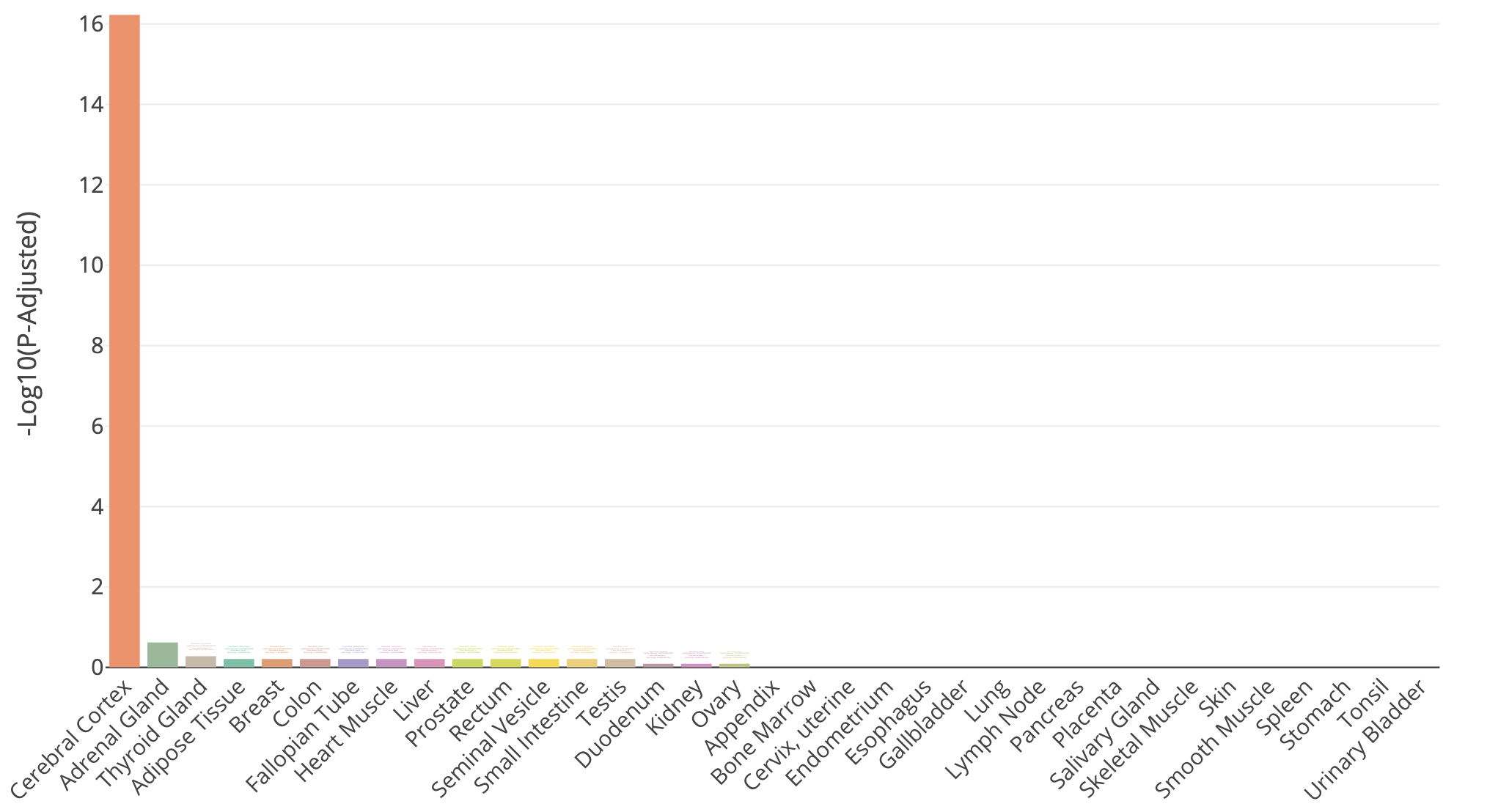
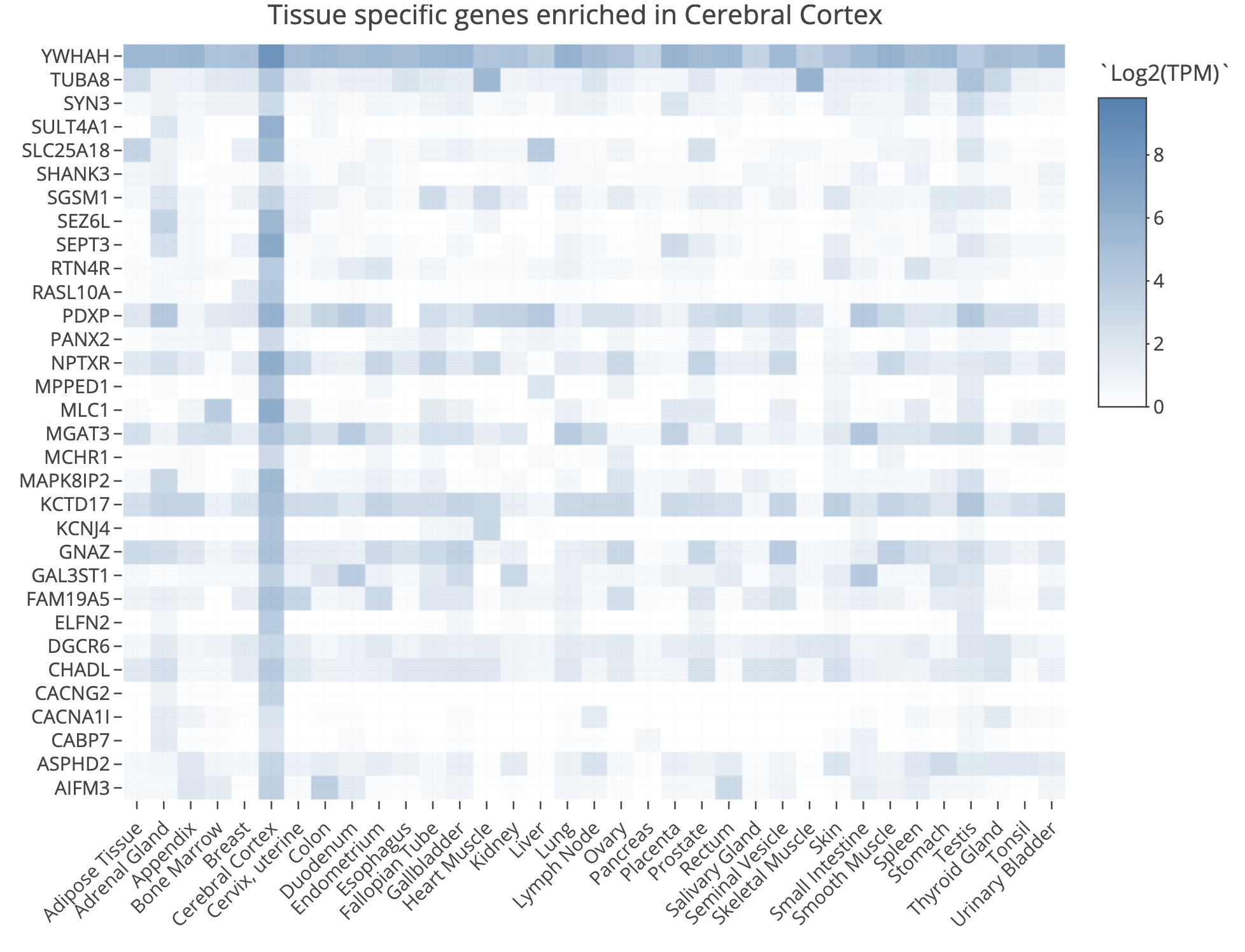
TissueEnrich result for Cerebral Cortex tissue
TissueEnrich example for UHR over-expressed gene: PRAME
TissueEnrich example for UHR over-expressed gene: SERPIND1
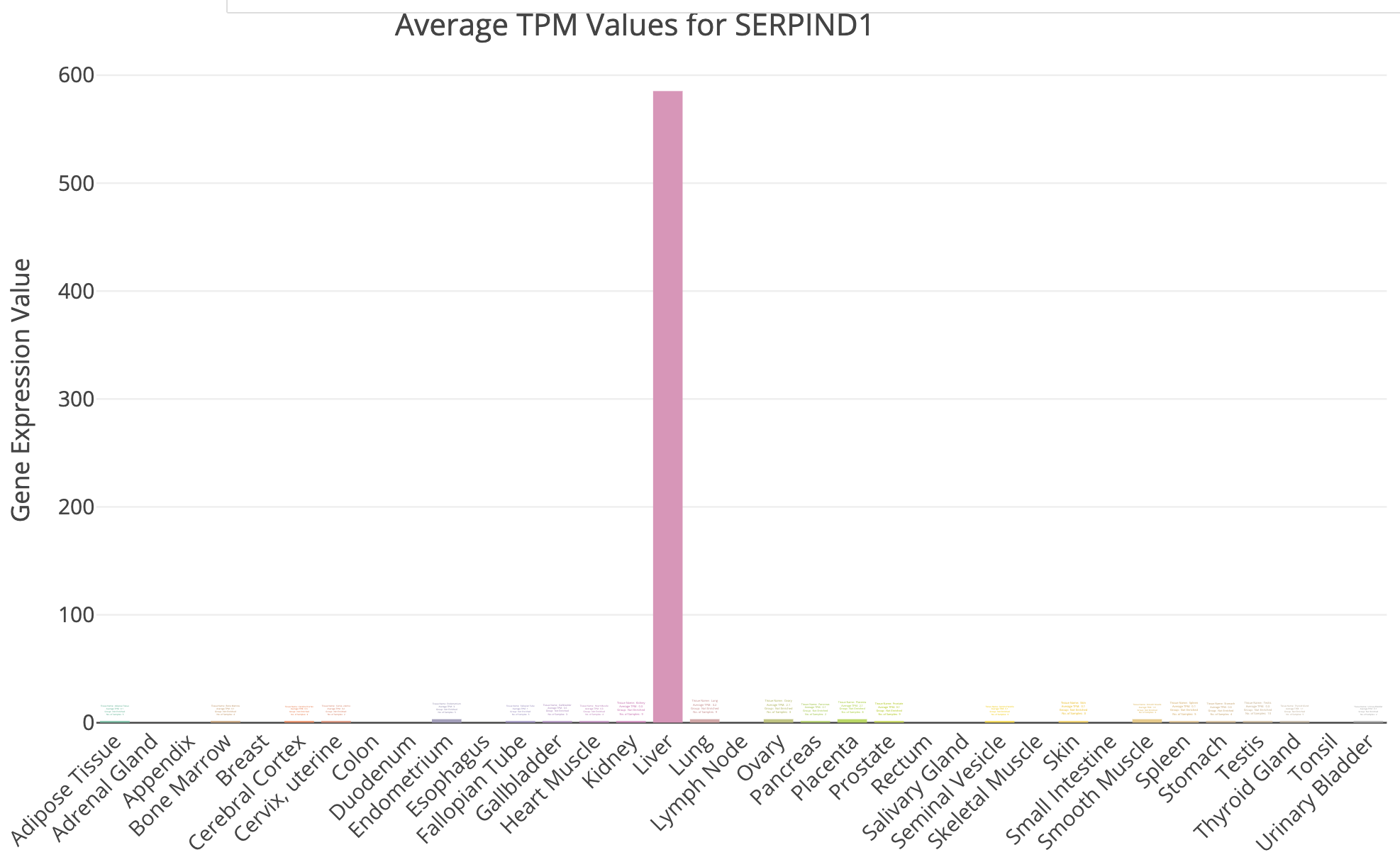
Does all of this make sense when we think about the makeup of the HBR and UHR samples? Refer back to the description of the samples.