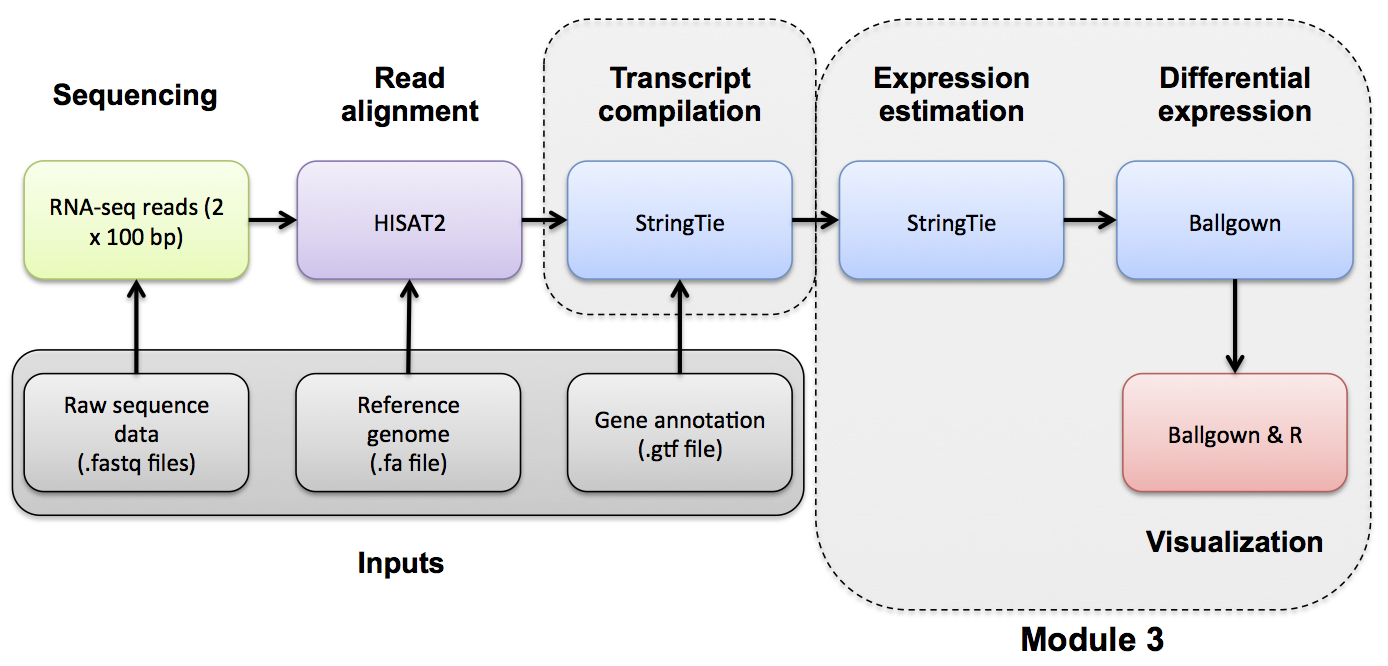
Differential Expression with Ballgown
Differential Expression mini lecture
If you would like a brief refresher on differential expression analysis, please refer to the mini lecture.
Ballgown DE Analysis
Use Ballgown to compare the UHR and HBR conditions. Refer to the Ballgown manual for a more detailed explanation:
Create and change to ballgown ref-only results directory:
mkdir -p $RNA_HOME/de/ballgown/ref_only/
cd $RNA_HOME/de/ballgown/ref_only/
Perform UHR vs. HBR comparison, using all replicates, for known (reference only mode) transcripts:
First, start an R session:
R
Run the following R commands in your R session.
# load the required libraries
library(ballgown)
library(genefilter)
library(dplyr)
library(devtools)
# Create phenotype data needed for ballgown analysis
ids = c("UHR_Rep1", "UHR_Rep2", "UHR_Rep3", "HBR_Rep1", "HBR_Rep2", "HBR_Rep3")
condition = c("UHR", "UHR", "UHR", "HBR", "HBR", "HBR")
inputs = "/home/ubuntu/workspace/rnaseq/expression/stringtie/ref_only/"
path = paste(inputs, ids, sep="")
pheno_data = data.frame(ids, condition, path)
# Load ballgown data structure and save it to a variable "bg"
bg = ballgown(samples = as.vector(pheno_data$path), pData = pheno_data)
# View the current order of the comparison: UHR/HBR (fold-changes > 1 means higher expression in UHR than HBR)
factor(pData(bg)$condition) # "Levels: HBR UHR", alphabetically by default determines the comparison
# Note that if you want to reverse the comparison of HBR and UHR. We would achieve this by changing the order of the "levels":
pData(bg)$condition <- factor(pData(bg)$condition, levels = c("UHR","HBR"))
factor(pData(bg)$condition) # "Levels: UHR HBR"
# Lets change the condition order back for consistency
pData(bg)$condition <- factor(pData(bg)$condition, levels = c("HBR","UHR"))
factor(pData(bg)$condition) # "Levels: HBR UHR"
# Display a description of this object
bg
# Load all attributes including gene name
bg_table = texpr(bg, 'all')
bg_gene_names = unique(bg_table[, 9:10])
bg_transcript_names = unique(bg_table[, c(1, 6)])
# Save the ballgown object to a file for later use
save(bg, file = 'bg.rda')
# Pull the gene and transcript expression data frame from the ballgown object
gene_expression = as.data.frame(gexpr(bg))
transcript_expression = as.data.frame(texpr(bg))
# Perform differential expression (DE) analysis with no filtering, at the transcript level
results_transcripts = stattest(bg, feature = "transcript", covariate = "condition", getFC = TRUE, meas = "FPKM")
# Then add on transcript/gene names and sample level fpkm values for export
results_transcripts = merge(results_transcripts, bg_transcript_names, by.x = c("id"), by.y = c("t_id"))
results_transcripts = merge(results_transcripts, transcript_expression, by.x = c("id"), by.y = c("row.names"))
# Perform differential expression (DE) analysis with no filtering, at the gene level
results_genes = stattest(bg, feature = "gene", covariate = "condition", getFC = TRUE, meas = "FPKM")
# Then add on transcript/gene names and sample level fpkm values for export
results_genes = merge(results_genes, bg_gene_names, by.x = c("id"), by.y = c("gene_id"))
results_genes = merge(results_genes, gene_expression, by.x = c("id"), by.y = c("row.names"))
# Note that in the statistical tests above, the design is very simple with condition as the single covariate.
# More complex design are possible including time series ("timecourse" parameter), confounders (using adjustvars parameter), etc.
# The ballgown vignettes describe these options in detail.
# Save a tab delimited file for both the transcript and gene results
write.table(results_transcripts, "UHR_vs_HBR_transcript_results.tsv", sep = "\t", quote = FALSE, row.names = FALSE)
write.table(results_genes, "UHR_vs_HBR_gene_results.tsv", sep = "\t", quote = FALSE, row.names = FALSE)
# Filter low-abundance genes. Here we remove all transcripts with a variance across the samples of less than one
bg_filt = subset (bg, "SparseArray::rowVars(texpr(bg)) > 1", genomesubset = TRUE)
# Load all attributes including gene name
bg_filt_table = texpr(bg_filt , 'all')
bg_filt_gene_names = unique(bg_filt_table[, 9:10])
bg_filt_transcript_names = unique(bg_filt_table[, c(1,6)])
# Perform DE analysis now using the filtered data
results_transcripts = stattest(bg_filt, feature = "transcript", covariate = "condition", getFC = TRUE, meas = "FPKM")
results_transcripts = merge(results_transcripts, bg_filt_transcript_names, by.x = c("id"), by.y = c("t_id"))
results_transcripts = merge(results_transcripts, transcript_expression, by.x = c("id"), by.y = c("row.names"))
results_genes = stattest(bg_filt, feature = "gene", covariate = "condition", getFC = TRUE, meas = "FPKM")
results_genes = merge(results_genes, bg_filt_gene_names, by.x = c("id"), by.y = c("gene_id"))
results_genes = merge(results_genes, gene_expression, by.x = c("id"), by.y = c("row.names"))
# Output the filtered list of genes and transcripts and save to tab delimited files
write.table(results_transcripts, "UHR_vs_HBR_transcript_results_filtered.tsv", sep = "\t", quote = FALSE, row.names = FALSE)
write.table(results_genes, "UHR_vs_HBR_gene_results_filtered.tsv", sep = "\t", quote = FALSE, row.names = FALSE)
# Identify the significant genes with p-value < 0.05
sig_transcripts = subset(results_transcripts, results_transcripts$pval<0.05)
sig_genes = subset(results_genes, results_genes$pval<0.05)
# Output the significant gene results to a pair of tab delimited files
write.table(sig_transcripts, "UHR_vs_HBR_transcript_results_sig.tsv", sep = "\t", quote = FALSE, row.names = FALSE)
write.table(sig_genes, "UHR_vs_HBR_gene_results_sig.tsv", sep = "\t", quote = FALSE, row.names = FALSE)
# Exit the R session
quit(save = "no")
Once you have completed the Ballgown analysis in R, exit the R session and continue with the steps below. A copy of the above R code is located here.
What does the raw output from Ballgown look like?
head UHR_vs_HBR_gene_results.tsv
How many genes are there on this chromosome?
grep -v feature UHR_vs_HBR_gene_results.tsv | wc -l
How many passed filter in UHR or HBR?
grep -v feature UHR_vs_HBR_gene_results_filtered.tsv | wc -l
How many differentially expressed genes were found on this chromosome (p-value < 0.05)?
grep -v feature UHR_vs_HBR_gene_results_sig.tsv | wc -l
Display the top 20 DE genes. Look at some of those genes in IGV - do they make sense?
In the following commands we use grep -v feature to remove lines that contain “feature”. Then we use sort to sort the data in various ways. The k option specifies that we want to sort on a particular column (3 in this case which has the DE fold change values). The n option tells sort to sort numerically. The r option tells sort to reverse the sort.
grep -v feature UHR_vs_HBR_gene_results_sig.tsv | sort -rnk 3 | head -n 20 | column -t #Higher abundance in UHR
grep -v feature UHR_vs_HBR_gene_results_sig.tsv | sort -nk 3 | head -n 20 | column -t #Higher abundance in HBR
Save all genes with P<0.05 to a new file.
grep -v feature UHR_vs_HBR_gene_results_sig.tsv | cut -f 6 | sed 's/\"//g' > DE_sig_genes_ballgown.tsv
head DE_sig_genes_ballgown.tsv
PRACTICAL EXERCISE 9
Assignment: Use Ballgown to identify differentially expressed genes from the StringTie expression estimates (i.e., Ballgown table files) which you created in Practical Exercise 8.
- Hint: Follow the example R code above.
- Hint: You will need to change how the
pheno_dataobject is created to point to the correct sample ids, condition, and path to your inputs (the StringTie results files). - Hint: Make sure to save your ballgown data object to file (e.g.,
bg.rda) for use in subsequent practical exercises. - Hint: You may wish to save both a complete list of genes with differential expression results as well as a subset which are filtered and pass a significance test
Solution: When you are ready you can check your approach against the Solutions
ERCC DE Analysis
This section will compare the differential expression estimates obtained by the RNAseq analysis against the expected differential expression results for the ERCC spike-in RNAs (mix1-UHR vs mix2-HBR):
First set up a directory to store the results of this analysis.
mkdir $RNA_HOME/de/ercc_spikein_analysis/
cd $RNA_HOME/de/ercc_spikein_analysis/
wget https://genomedata.org/rnaseq-tutorial/ERCC_Controls_Analysis.txt
cat ERCC_Controls_Analysis.txt
Using R load the expected and observed ERCC DE results and produce a visualization.
First, start an R session:
R
Work through the following R commands
library(ggplot2)
#load the ERCC expected fold change values for mix1 vs mix2
ercc_ref = read.table("ERCC_Controls_Analysis.txt", header=TRUE, sep="\t")
names(ercc_ref) = c("id", "ercc_id", "subgroup", "ref_conc_mix_1", "ref_conc_mix_2", "ref_fc_mix1_vs_mix2", "ref_log2_mix1_vs_mix2")
head(ercc_ref)
dim(ercc_ref)
#load the observed fold change values determined by our RNA-seq analysis
rna_de_file = "~/workspace/rnaseq/de/ballgown/ref_only/UHR_vs_HBR_gene_results.tsv";
rna_de = read.table(rna_de_file, header=TRUE, sep="\t")
tail(rna_de)
dim(rna_de)
#combine the expected and observed data into a single data table
ercc_ref_de = merge(x = ercc_ref, y = rna_de, by.x = "ercc_id", by.y = "id", all.x = TRUE)
head(ercc_ref_de)
dim(ercc_ref_de)
#convert fold change values to log2 scale
ercc_ref_de$observed_log2_fc = log2(ercc_ref_de$fc)
ercc_ref_de$expected_log2_fc = ercc_ref_de$ref_log2_mix1_vs_mix2
#fit a linear model and calculate R squared between the observed and expected fold change values
model = lm(observed_log2_fc ~ expected_log2_fc, data=ercc_ref_de)
r_squared = summary(model)[["r.squared"]]
#create a scatterplot to compare the observed and expected fold change values
p = ggplot(ercc_ref_de, aes(x = expected_log2_fc, y = observed_log2_fc))
p = p + geom_point(aes(color = subgroup))
p = p + geom_smooth(method = lm)
p = p + annotate("text", 1, 2, label=paste("R^2 =", r_squared, sep=" "))
p = p + xlab("Expected Fold Change (log2 scale)")
p = p + ylab("Observed Fold Change in RNA-seq data (log2 scale)")
#save the plot to a PDF
pdf("ERCC_Ballgown-DE_vs_SpikeInDE.pdf")
print(p)
dev.off()
# Exit the R session
quit(save="no")
View the results here:
- https://YOUR_PUBLIC_IPv4_ADDRESS/rnaseq/de/ercc_spikein_analysis/ERCC_Ballgown-DE_vs_SpikeInDE.pdf