Posts
My thoughts and ideas
Welcome to the blog
My thoughts and ideas
Introduction to bioinformatics for RNA sequence analysis
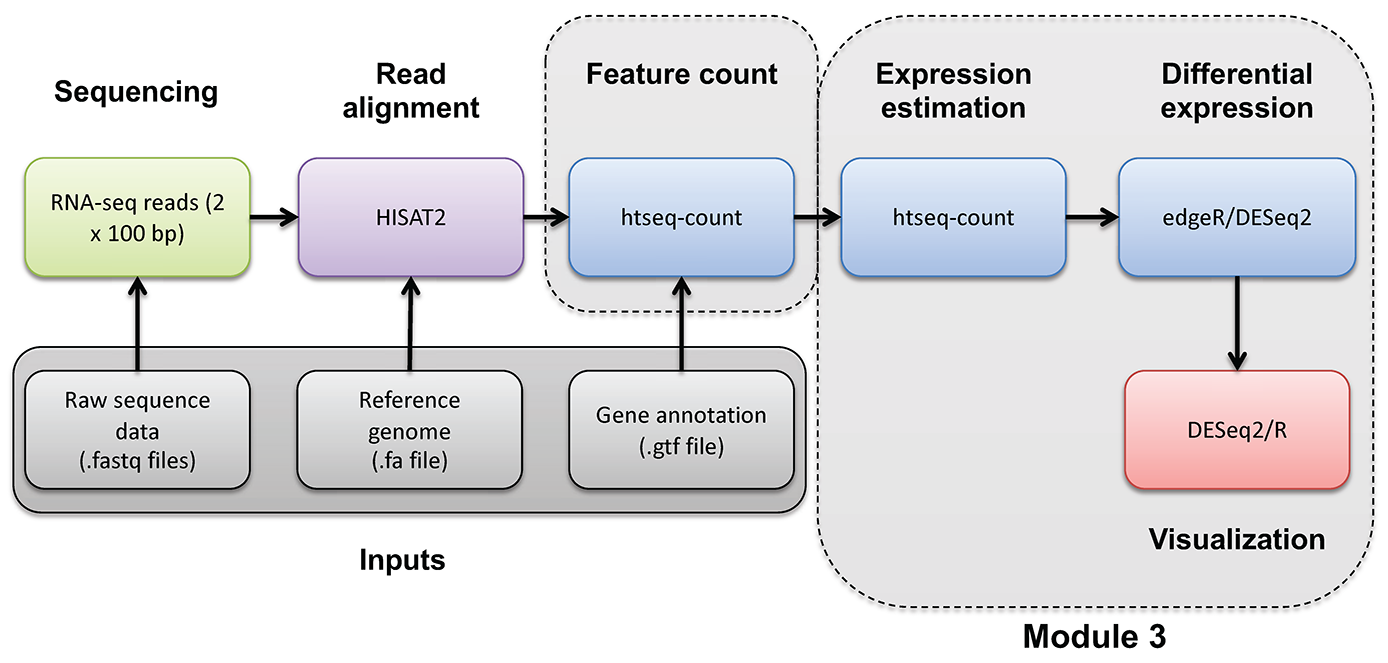
In this section we will obtain a dataset to allow demonstration of batch correction using the ComBat-Seq tool in R (Bioconductor).
For this exercise we will obtain public RNA-seq data from an extensive multi-platform comparison of sequencing platforms that also examined the impact of generating data at multiple sites, using polyA vs ribo-reduction for enrichment, and the impact of RNA degradation (PMID: 25150835): “Multi-platform and cross-methodological reproducibility of transcriptome profiling by RNA-seq in the ABRF Next-Generation Sequencing Study”.
This publication used the same UHR (cancer cell lines) and HBR (brain tissue) samples we have been using throughout this course. To examine a strong batch effect, we will consider a DE analysis of UHR vs HBR where we compare Ribo-depleted (“Ribo”) and polyA-enriched (“Poly”) samples.
The entire RNA-seq dataset PMID: 25150835 used for this module has been deposited in GEO. In GEO, these data are organized as a superseries: GSE46876 which has data for several sequencing platforms. The data from the Illumina Platform are part of this subseries: GSE48035.
To do this analysis quickly, we will download pre-computed raw read counts for this dataset: GSE48035_ILMN.counts.txt.gz
Set up a working directory and download the RNA-seq counts file needed for the following exercise as follows:
cd $RNA_HOME
mkdir batch_correction
cd batch_correction
wget https://genomedata.org/rnaseq-tutorial/batch_correction/GSE48035_ILMN.counts.txt.gz
Create a simplified version of this file that has only the counts for the samples we wish to use for this analysis as follows:
cd $RNA_HOME/batch_correction
#remove all quotes from file
zcat GSE48035_ILMN.counts.txt.gz | tr -d '"' > GSE48035_ILMN.counts.tmp.txt
#create a fixed version of the header and store for later
(echo -e "Gene\tChr\t"; head -n 1 GSE48035_ILMN.counts.tmp.txt) | tr -d '\n' > header.txt
#split the chromosome and gene names on each line, sort the file by gene name
#ensure input and output are interpreted as tab separated.
#Split the first entry on "!", replace with a new first entry the reverses chr and gene name
awk -F'\t' 'BEGIN { OFS="\t" } {
split($1, a, "!");
$1 = a[2] OFS a[1];
print
}' GSE48035_ILMN.counts.tmp.txt | sort > GSE48035_ILMN.counts.tmp2.txt
#remove the old header line
grep -v --color=never ABRF GSE48035_ILMN.counts.tmp2.txt > GSE48035_ILMN.counts.clean.txt
#cut out only the columns for the UHR (A) and HBR (B) samples, replicates 1-4, and PolyA vs Enrichment
cut -f 1-2,3-6,7-10,19-22,23-26 GSE48035_ILMN.counts.clean.txt > GSE48035_ILMN.Counts.SampleSubset.txt
cut -f 1-2,3-6,7-10,19-22,23-26 header.txt > header.SampleSubset.txt
#how many gene lines are we starting with?
wc -l GSE48035_ILMN.Counts.SampleSubset.txt
#cleanup intermediate files created above
rm -f GSE48035_ILMN.counts.txt.gz GSE48035_ILMN.counts.tmp.txt GSE48035_ILMN.counts.tmp2.txt GSE48035_ILMN.counts.clean.txt header.txt
Further limit these counts to those that correspond to known protein coding genes:
cd $RNA_HOME/batch_correction
#download complete Ensembl GTF file
wget ftp://ftp.ensembl.org/pub/release-101/gtf/homo_sapiens/Homo_sapiens.GRCh38.101.gtf.gz
#grab all the gene records, limit to gene with "protein_coding" biotype, create unique gene name list
zcat Homo_sapiens.GRCh38.101.gtf.gz | grep -w gene | grep "gene_biotype \"protein_coding\"" | cut -f 9 | cut -d ";" -f 3 | tr -d " gene_name " | tr -d '"' | sort | uniq > Ensembl101_ProteinCodingGeneNames.txt
#how many unique protein coding genes names does Ensembl have?
wc -l Ensembl101_ProteinCodingGeneNames.txt
#filter our gene count matrix down to only the protein coding genes
join -j 1 -t $'\t' Ensembl101_ProteinCodingGeneNames.txt GSE48035_ILMN.Counts.SampleSubset.txt | cat header.SampleSubset.txt - > GSE48035_ILMN.Counts.SampleSubset.ProteinCodingGenes.tsv
#how many lines of RNA-seq counts do we still have?
wc -l GSE48035_ILMN.Counts.SampleSubset.ProteinCodingGenes.tsv
#clean up
rm -f header.SampleSubset.txt GSE48035_ILMN.Counts.SampleSubset.txt
#take a look at the final filtered read count matrix to be used for the following analysis
column -t GSE48035_ILMN.Counts.SampleSubset.ProteinCodingGenes.tsv | less -S
Note that filtering gene lists by gene name as we have done above is generally not advised as we usually can’t guarantee that gene names from two different lists are compatible. Mapping between unique identifiers would be preferable. But for demonstrating the batch analysis below this should be fine…