Posts
My thoughts and ideas
Welcome to the blog
My thoughts and ideas
Introduction to bioinformatics for RNA sequence analysis
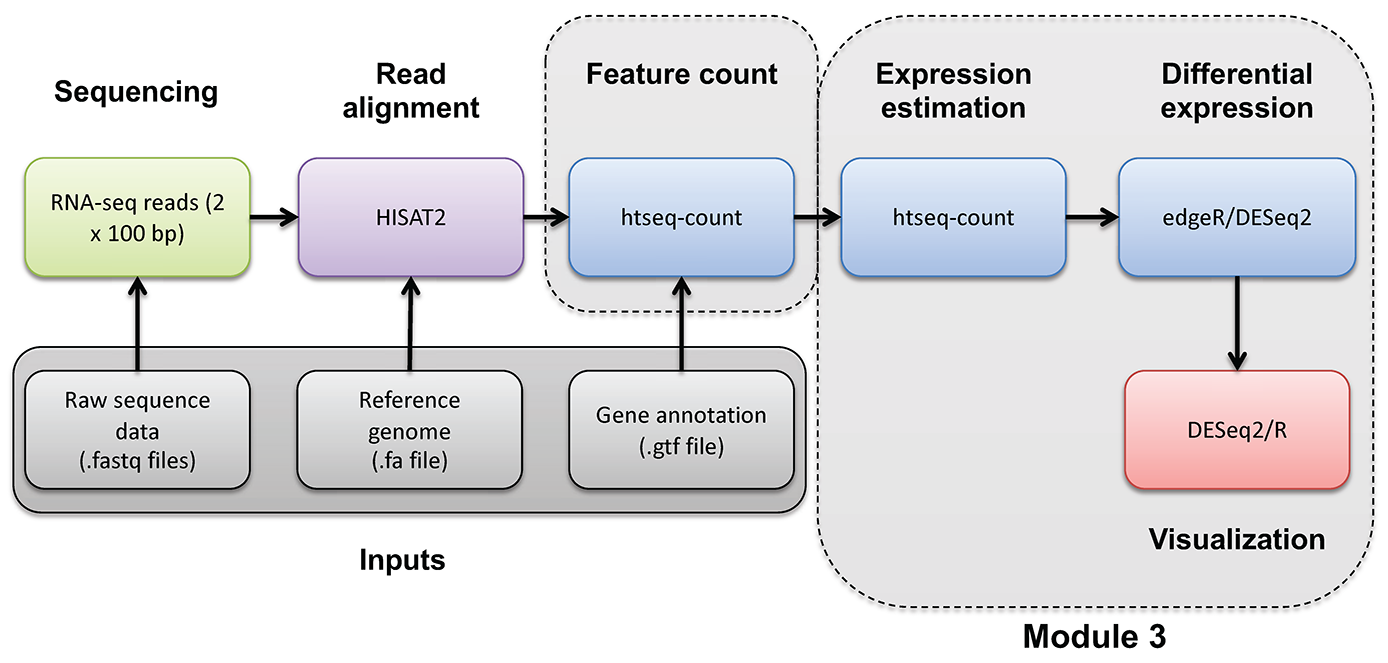
In this tutorial you will:
We will use a google form to capture basic information about AI tool choice (and model version), AI prompts used, etc. Refer to the course slack channel for a link to this form.
For this exercise we want you to imagine that you did not have the previous section as a guide on how to perform a differential expression analysis in R. Imagine that you obtained the gene counts matrix “gene_read_counts_table_all_final.tsv” from your sequencing core, a collaborator, or a public source. See if you can complete a DE analysis like the one you just completed, step-by-step, but using an AI assistant. Here is an overview of the basic steps (more details on each below):
For the exercise use the following:
/home/ubuntu/workspace/rnaseq/expression/htseq_counts/gene_read_counts_table_all_final.tsv
/home/ubuntu/workspace/rnaseq/de/ENSG_ID2Name.txt.gz
/home/ubuntu/workspace/rnaseq/de_ai_exercise
Hint. To quickly see what the input data looks like, you can do the following to display the first two lines of each:
head(read.delim("/home/ubuntu/workspace/rnaseq/expression/htseq_counts/gene_read_counts_table_all_final.tsv"), n=2)
head(read.delim("/home/ubuntu/workspace/rnaseq/de/ENSG_ID2Name.txt"), n=2)
There are several broad approaches to using AI for bioinformatics analysis, and your choice of tool will shape how the interaction unfolds. The main paradigms are:
Conversational AI assistants. You interact with the AI through a chat interface, describing your task in plain language and exchanging messages back and forth. You paste in data snippets or describe your files, and the AI generates code or explanations in response. This is the most accessible entry point and requires no special software setup.
Integrated development environment (IDE) and command line interface (CLI) AI coding tools. Some development environments embed AI assistance directly into the coding workflow. The AI can see your open files, suggest completions inline, explain errors in context, and generate code without leaving the editor or commandline. This tighter integration can make iteration faster but requires a compatible editor/tool.
Locally-run open models. Open-weight language models can be run on your own machine, which avoids sending data to external servers (relevant when working with sensitive or unpublished data). These require more setup and capable hardware, and are generally less capable than leading commercial models at time of writing.
Note that given the cloud-based R/RStudio environment used in this course, the conversational assistant approach is the most practical. Using this approach you can work in a browser tab alongside your R or RStudio session with no additional setup. The IDE-integrated approach is possible but would require downloading the input data files to your laptop. Locally-run models are beyond the scope of this exercise.
For this exercise, use whichever AI tool you are comfortable with, or take the opportunity to try one you have heard about but not used before, or simply pick one from the list below. A few commonly used options across these categories:
| Tool | Type | Provider |
|---|---|---|
| ChatGPT | Conversational | OpenAI |
| Claude | Conversational | Anthropic |
| Gemini | Conversational | |
| Microsoft Copilot | Conversational | Microsoft |
| GitHub Copilot | IDE-based | Microsoft/GitHub |
| Cursor | IDE-based | Cursor |
| Claude Code | CLI-based | Anthropic |
| Codex | CLI-based | OpenAI |
| Ollama | Local models | Meta (open weight) |
| Qwen | Local models | Alibaba (open weight) |
| DeepSeek | Local models | deepseek (open weight) |
Make note of both the tool and the specific model version you use (e.g. GPT-4o, Claude Sonnet 3.7, Gemini 2.0 Flash). Model versions vary significantly in capability and the field moves quickly. Record your choice in the Google form.
Before starting, take a few minutes to think about what you will tell the AI. A well-constructed prompt is likely to produce more useful code than a vague one, and the differences across students will make for a rich comparison later. You do not need to craft a perfect prompt! Part of the point of this exercise is to see what happens with different approaches.
As you draft your prompt, consider what information the AI would need to produce useful, runnable code. Some dimensions to think about (without being told exactly what to include):
Write your initial prompt, then record it in the Google form before sending it to the AI. You may refine it through the conversation.
Enter your prompt into the AI tool of your choice and examine the response carefully before running anything. Consider:
Iterate with the AI as needed. When you are satisfied, copy the code into your cloud R environment and execute it line by line. At each step, inspect the output and try to understand what the code is doing before moving on. If something fails or produces unexpected results, you may go back to the AI to debug.
Record your answers to the above questions, along with any significant follow-up prompts, in the Google form.
Before wrapping up, make sure your analysis produces results that address the following. These are the questions you will answer in the Google form and the basis for class discussion:
Record your answers in the Google form and save your volcano plot image to your results directory. Be prepared to share it for class discussion. The variation across students in both results and plot appearance will be part of what we examine together.
The following resources are starting points for deepening your understanding of the concepts touched on in this exercise. This is a fast-moving area. Treat these as entry points rather than definitive answers.
Comparing and choosing AI tools
Prompt engineering
Critical evaluation and responsible use
AI tools for bioinformatics and data science