DE Visualization with DESeq2
Differential Expression Visualization
In this section we will be going over some basic visualizations of the DESeq2 results generated in the “Differential Expression with DESeq2” section of this course. Our goal is to quickly obtain some interpretable results using built-in visualization functions from DESeq2 or recommended packages. For a very extensive overview of DESeq2 and how to visualize and interpret the results, refer to the DESeq2 vignette.
Setup
Navigate to the correct directory and then launch R:
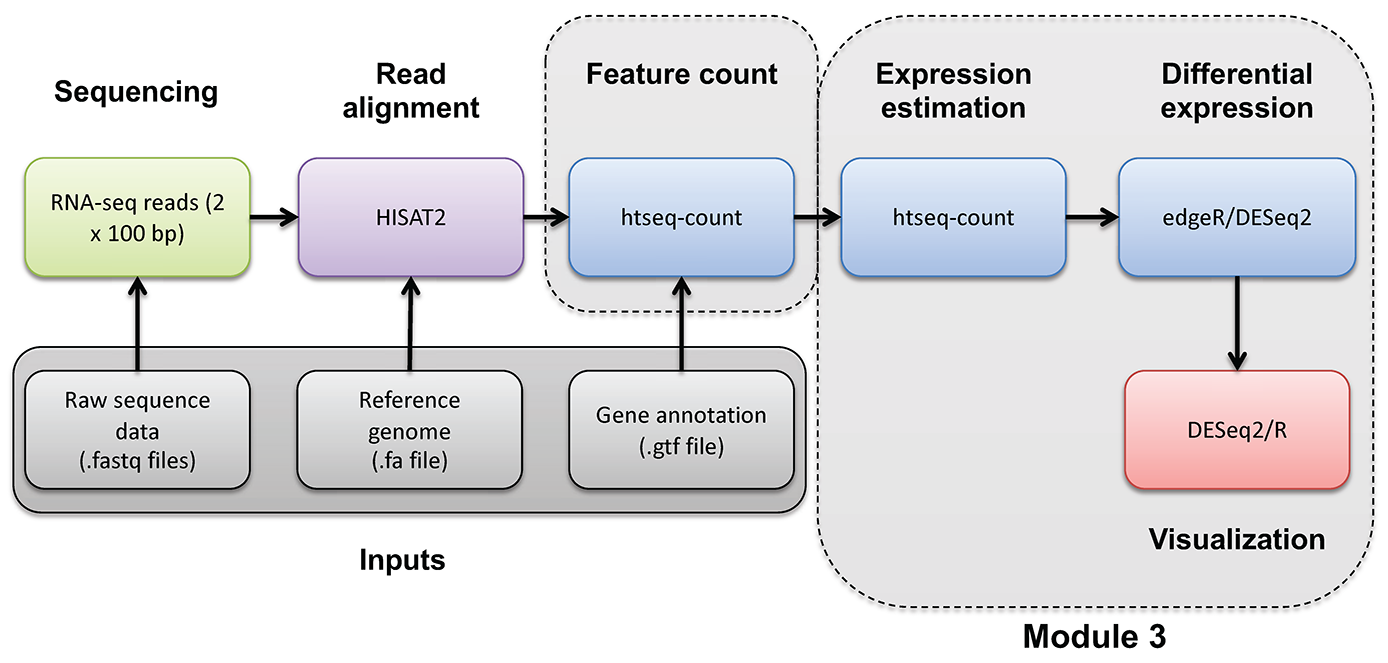
cd $RNA_HOME/de/htseq_counts/deseq2
R
If it is not already in your R environment, load the DESeqDataSet object and the results table into the R environment.
# set the working directory
setwd("/home/ubuntu/workspace/rnaseq/de/htseq_counts/deseq2")
# view the contents of this directory
dir()
# load libs
library(DESeq2)
library(data.table)
library(pheatmap)
# Load in the DESeqDataSet object (http://genomedata.org/cri-workshop/deseq2/dds.rds)
dds = readRDS("dds.rds")
# Load in the results object before shrinkage (http://genomedata.org/cri-workshop/deseq2/res.rds)
res = readRDS("res.rds")
# Load in the results object after shrinkage (http://genomedata.org/cri-workshop/deseq2/resLFC.rds)
resLFC = readRDS("resLFC.rds")
# Load in the final results file with all sorted DE results (http://genomedata.org/cri-workshop/deseq2/DE_all_genes_DESeq2.tsv)
deGeneResultSorted = fread("DE_all_genes_DESeq2.tsv")
MA-plot before LFC shrinkage
MA-plots were originally used to evaluate microarray expression data where M is the log ratio and A is the mean intensity for a gene (both based on scanned intensity measurements from the microarray).
These types of plots are still usefull in RNAseq DE experiments with two conditions, as they can immediately give us information on the number of signficantly differentially expressed genes, the ratio of up vs down regulated genes, and any outliers. To interpret these plots it is important to keep a couple of things in mind. The Y axis (M) is the log2 fold change between the two conditions tested, a higher fold-change indicates greater difference between condition A and condition B. The X axis (A) is a measure of read alignment to a gene, so as you go higher on on the X axis you are looking at regions which have higher totals of aligned reads, in other words the gene is “more” expressed overall (with the caveat that gene length is not being taken into account by raw read counts here).
Using the built-in plotMA function from DESeq2 we also see that the genes are color coded by a significance threshold (e.g., adjusted p-value < 0.1). Genes with higher expression values and higher fold-changes are more often significant as one would expect.
})
# use DESeq2 built in MA-plot function
pdf("maplot_preShrink.pdf")
plotMA(res, alpha = 0.1, ylim=c(-2, 2), cex=1.5, main = "MA-plot before LFC shrinkage")
dev.off()
MA-plot after LFC shrinkage
When we ran DESeq2 we obtained two results, one with and one without log-fold change shrinkage. When you have genes with low hits you can get some very large fold changes. For example 1 hit on a gene vs 6 hits on a gene is a 6x fold change. This high level of variance though is probably quantifying noise instead of real biology. Running plotMA on our results where we applied an algorithm for log fold change shrinkage we can see that this “effect” is somewhat controlled for. I do want to note that while shrinking LFC is part of a typical DE workflow there are cases where you would not want to perform this, namely when there is already low variation amongst samples (i.e. from technical replicates) as most shrinkage algorithms rely on some variability to build a prior distribution.
# use DESeq2 built in MA-plot function
pdf("maplot_postShrink.pdf")
plotMA(resLFC, alpha = 0.1, ylim = c(-2, 2), cex=1.5, main = "MA-plot after LFC shrinkage")
dev.off()
The effect is very subtle here due to the focused nature of our dataset (chr22 genes only), but if you toggle between the two plots and look in the upper left and bottom left corners you can see some fold change values are moving closer to 0.
Viewing individual gene counts between two conditions
Often it may be useful to view the normalized counts for a gene amongst our samples. DESeq2 provides a built in function for that which works off of the dds object. Here we view SEPT3 which we can see in our DE output is significantly higher in the HBR cohort and PRAME which is significantly higher in the UHR cohort. This is useful as we can see the per-sample distribution of our corrected counts, we can immediately determine if there are any outliers within each group and investigate further if need be.
# hint! you defined intgroup when creating the dds object, you can view the name by printing the dds objct in your R session
# dds
pdf("normalized_count_examples.pdf")
# view SEPT3 normalized counts
plotCounts(dds, gene = "ENSG00000100167", intgroup = "Condition", main = "SEPT3")
# view PRAME normalized counts
plotCounts(dds, gene = "ENSG00000185686", intgroup = "Condition", main = "PRAME")
dev.off()
Viewing pairwise sample clustering
It may often be useful to view inter-sample relatedness. In other words, how similar or disimilar samples are to one another overall. While not part of the DESeq2 package, there is a convenient library that can easily construct a hierarchically clustered heatmap from our DESeq2 data. It should be noted that when doing a distance calculation using “raw count” data is not ideal, the count data should be transformed using vst() or rlog() which can be performed directly on the dds object. The reason for this is described in detail in the DESeq2 manuscript, suffice it to say that transforming gene variance to be more homoskedastic will make inferences of sample relatedness more interpretable.
# note that we use rlog because we don't have a large number of genes, for a typical DE experiment with 1000's of genes use the vst() function
rld <- rlog(dds, blind = FALSE)
# view the structure of this object
rld
# compute sample distances (the dist function uses the euclidean distance metric by default)
# in this command we will pull the rlog transformed data ("regularized" log2 transformed, see ?rlog for details) using "assay"
# then we transpose that data using t()
# then we calculate distance values using dist()
# the distance is calculated for each vector of sample gene values, in a pairwise fashion comparing all samples
# view the first few lines of raw data
head(assay(dds))
# see the rlog transformed data
head(assay(rld))
# see the impact of transposing the matrix
t(assay(rld))[1:6, 1:5]
# see the distance values
dist(t(assay(rld)))
# put it all together and store the result
sampleDists <- dist(t(assay(rld)))
# convert the distance result to a matrix
sampleDistMatrix <- as.matrix(sampleDists)
# view the distance numbers directly in the pairwise distance matrix
head(sampleDistMatrix)
pdf("distance_sample_heatmap.pdf", width = 8, height = 8)
# construct clustered heatmap, important to use the computed sample distances for clustering
pheatmap(sampleDistMatrix, clustering_distance_rows = sampleDists, clustering_distance_cols = sampleDists)
dev.off()
Instead of a distance metric we could also use a similarity metric such as a Peason correlation
There are many correlation and distance options:
- Similarity metrics: “pearson”, “kendall”, “spearman”
- Distance metrics: “euclidean”, “maximum”, “manhattan”, “canberra”, “binary” or “minkowski”
sampleCorrs = cor(assay(rld), method = "pearson")
sampleCorrMatrix = as.matrix(sampleCorrs)
head(sampleCorrMatrix)
pdf("similarity_sample_heatmap.pdf", width = 8, height = 8)
pheatmap(sampleCorrMatrix)
dev.off()
Instead of boiling all the gene count data for each sample down to a distance metric you can get a similar sense of the pattern by just visualizing all the genes and their expression at once
pdf("all_gene_heatmap.pdf", width = 10, height = 10)
# because there are so many gene we choose not to display them
pheatmap(mat = t(assay(rld)), show_colnames = FALSE)
dev.off()
Make a heatmap for just significant genes (e.g, FC >=3 and padj<0.001). Note, we chose these cutoffs arbitrarily to result in a number of genes that can be legibly labeled
#First, get a list of just significant genes
deGeneSigResult=deGeneResultSorted[(deGeneResultSorted$padj<0.001 & (deGeneResultSorted$log2FoldChange>=3 | deGeneResultSorted$log2FoldChange<=-3)),]
SigGenes=deGeneSigResult$ensemblID
SigGeneSymbols=deGeneSigResult$Symbol
#extract expression data
expression_data=assay(rld)
#Limit to significant genes
expression_data=expression_data[SigGenes,]
#Create a heatmap as before
pdf("sig_gene_heatmap.pdf", width = 10, height = 10)
pheatmap(mat = t(expression_data), show_colnames = TRUE, labels_col=SigGeneSymbols)
dev.off()
quit(save="no")