Adapter Trim
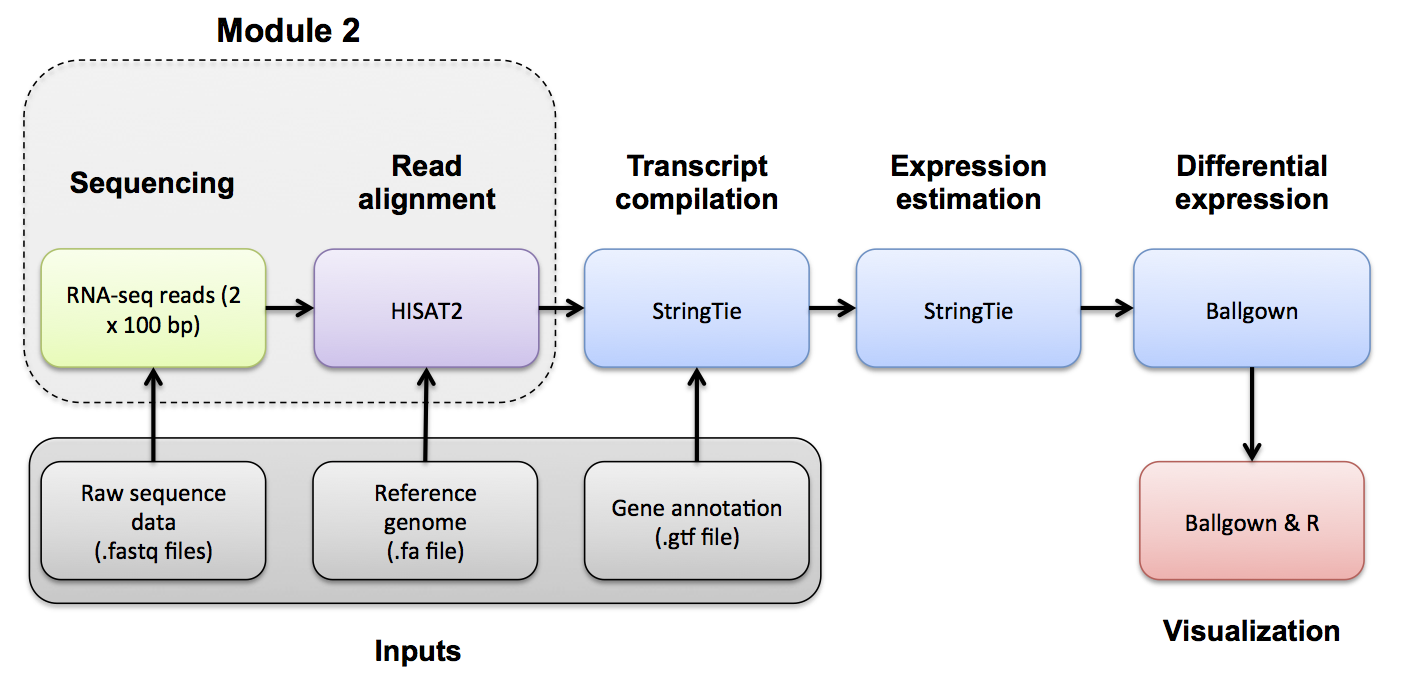
[OPTIONAL]
Use Fastp to trim sequence adapter from the read FASTQ files and also perform basic data quality cleanup. The output of this step will be trimmed and filtered FASTQ files for each data set.
Refer to the Fastp project and manual for a more detailed explanation:
Fastp basic usage:
fastp -i in.R1.fq.gz -I in.R2.fq.gz -o out.R1.fq.gz -O out.R2.fq.gz
The -i and -I parameters specify input R1 and R2 data files (raw data)
The -o and -O parameters specify output R1 and R2 data files (trimmed and quality filtered)
Extra options specified below:
- ‘-l 25’ the minimum read length allowed after trimming is 25bp
- ’–adapter_fasta’ the path to the adapter FASTA file containing adapter sequences to trim
- ’–trim_front1 13’ trim a fixed number (13 in this case) of bases off the left end of read1
- ’–trim_front2 13’ trim a fixed number (13 in this case) of bases off the left end of read2
- ’–json’ the path to store a log file in JSON file format
- ’–html’ the path to store a web report file
- ‘2>’ use to store the information that would be printed to the screen into a file instead
Read trimming with Fastp
First, set up some directories for output
echo $RNA_DATA_TRIM_DIR
mkdir -p $RNA_DATA_TRIM_DIR
Download necessary Illumina adapter sequence files.
echo $RNA_REFS_DIR
mkdir -p $RNA_REFS_DIR
cd $RNA_REFS_DIR
wget https://genomedata.org/rnaseq-tutorial/illumina_multiplex.fa
Use fastp to remove illumina adapter sequences (if any), trim the first 13 bases of each read, and perform default read quality filtering to remove reads that are too short, have too many low quality bases or have too many N’s.
cd $RNA_HOME
export S1=UHR_Rep1_ERCC-Mix1_Build37-ErccTranscripts-chr22
fastp -i $RNA_DATA_DIR/${S1}.read1.fastq.gz -I $RNA_DATA_DIR/${S1}.read2.fastq.gz -o $RNA_DATA_TRIM_DIR/${S1}.read1.fastq.gz -O $RNA_DATA_TRIM_DIR/${S1}.read2.fastq.gz -l 25 --adapter_fasta $RNA_REFS_DIR/illumina_multiplex.fa --trim_front1 13 --trim_front2 13 --json $RNA_DATA_TRIM_DIR/${S1}.fastp.json --html $RNA_DATA_TRIM_DIR/${S1}.fastp.html 2>$RNA_DATA_TRIM_DIR/${S1}.fastp.log
export S2=UHR_Rep2_ERCC-Mix1_Build37-ErccTranscripts-chr22
fastp -i $RNA_DATA_DIR/${S2}.read1.fastq.gz -I $RNA_DATA_DIR/${S2}.read2.fastq.gz -o $RNA_DATA_TRIM_DIR/${S2}.read1.fastq.gz -O $RNA_DATA_TRIM_DIR/${S2}.read2.fastq.gz -l 25 --adapter_fasta $RNA_REFS_DIR/illumina_multiplex.fa --trim_front1 13 --trim_front2 13 --json $RNA_DATA_TRIM_DIR/${S2}.fastp.json --html $RNA_DATA_TRIM_DIR/${S2}.fastp.html 2>$RNA_DATA_TRIM_DIR/${S2}.fastp.log
export S3=UHR_Rep3_ERCC-Mix1_Build37-ErccTranscripts-chr22
fastp -i $RNA_DATA_DIR/${S3}.read1.fastq.gz -I $RNA_DATA_DIR/${S3}.read2.fastq.gz -o $RNA_DATA_TRIM_DIR/${S3}.read1.fastq.gz -O $RNA_DATA_TRIM_DIR/${S3}.read2.fastq.gz -l 25 --adapter_fasta $RNA_REFS_DIR/illumina_multiplex.fa --trim_front1 13 --trim_front2 13 --json $RNA_DATA_TRIM_DIR/${S3}.fastp.json --html $RNA_DATA_TRIM_DIR/${S3}.fastp.html 2>$RNA_DATA_TRIM_DIR/${S3}.fastp.log
export S4=HBR_Rep1_ERCC-Mix2_Build37-ErccTranscripts-chr22
fastp -i $RNA_DATA_DIR/${S4}.read1.fastq.gz -I $RNA_DATA_DIR/${S4}.read2.fastq.gz -o $RNA_DATA_TRIM_DIR/${S4}.read1.fastq.gz -O $RNA_DATA_TRIM_DIR/${S4}.read2.fastq.gz -l 25 --adapter_fasta $RNA_REFS_DIR/illumina_multiplex.fa --trim_front1 13 --trim_front2 13 --json $RNA_DATA_TRIM_DIR/${S4}.fastp.json --html $RNA_DATA_TRIM_DIR/${S4}.fastp.html 2>$RNA_DATA_TRIM_DIR/${S4}.fastp.log
export S5=HBR_Rep2_ERCC-Mix2_Build37-ErccTranscripts-chr22
fastp -i $RNA_DATA_DIR/${S5}.read1.fastq.gz -I $RNA_DATA_DIR/${S5}.read2.fastq.gz -o $RNA_DATA_TRIM_DIR/${S5}.read1.fastq.gz -O $RNA_DATA_TRIM_DIR/${S5}.read2.fastq.gz -l 25 --adapter_fasta $RNA_REFS_DIR/illumina_multiplex.fa --trim_front1 13 --trim_front2 13 --json $RNA_DATA_TRIM_DIR/${S5}.fastp.json --html $RNA_DATA_TRIM_DIR/${S5}.fastp.html 2>$RNA_DATA_TRIM_DIR/${S5}.fastp.log
export S6=HBR_Rep3_ERCC-Mix2_Build37-ErccTranscripts-chr22
fastp -i $RNA_DATA_DIR/${S6}.read1.fastq.gz -I $RNA_DATA_DIR/${S6}.read2.fastq.gz -o $RNA_DATA_TRIM_DIR/${S6}.read1.fastq.gz -O $RNA_DATA_TRIM_DIR/${S6}.read2.fastq.gz -l 25 --adapter_fasta $RNA_REFS_DIR/illumina_multiplex.fa --trim_front1 13 --trim_front2 13 --json $RNA_DATA_TRIM_DIR/${S6}.fastp.json --html $RNA_DATA_TRIM_DIR/${S6}.fastp.html 2>$RNA_DATA_TRIM_DIR/${S6}.fastp.log
Use FastQC and multiqc to compare the impact of trimming
Optional exercise: Compare the FastQC reports for fastq files before and after trimming. All fastqc reports can be generated on the commandline.
cd $RNA_DATA_TRIM_DIR
fastqc *.fastq.gz
multiqc ./
The resulting html reports can be viewed by navigating to:
- https://YOUR_PUBLIC_IPv4_ADDRESS/rnaseq/data/
- https://YOUR_PUBLIC_IPv4_ADDRESS/rnaseq/data/trimmed/
Clean up
Move the fastqc and fastp results into sub-directories to keep things tidy
cd $RNA_DATA_TRIM_DIR
mkdir fastqc
mv *_fastqc* fastqc
mkdir fastp
mv *fastp.* fastp
PRACTICAL EXERCISE 5
Assignment: Using the approach above, trim the reads for both normal and tumor samples that you downloaded for the previous practical exercise. NOTE: try dropping the hard left trim option used above (‘–trim_front1 13’ and ‘–trim_front2 13’). Once you have trimmed the reads, compare a pre- and post- trimming FastQ file using the FastQC and multiqc tools.
- Hint: These files should have been downloaded to $RNA_HOME/practice/data/.
Questions
Answer these questions by examining the FastQC reports:
- After trimming, what is the range of read lengths observed for hcc1395 normal replicate 1, read 1?
- Which sections of the FastQC report are most informative for observing the effect of trimming?
- In the ‘Per base sequence content section’, what pattern do you see? What could explain this pattern?
Solution: When you are ready you can check your approach against the Solutions.