RNAseq Data
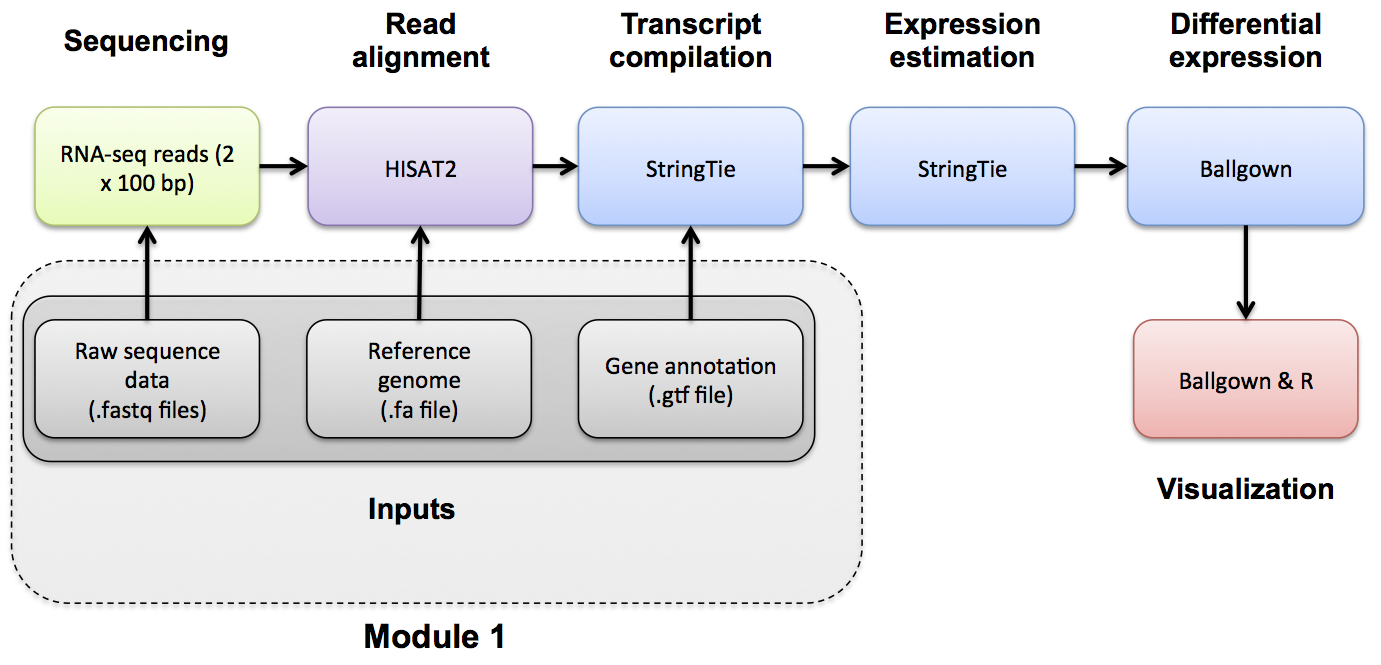
Obtain RNA-seq test data.
The test data consists of two commercially available RNA samples: Universal Human Reference (UHR) and Human Brain Reference (HBR). The UHR is total RNA isolated from a diverse set of 10 cancer cell lines (breast, liver, cervix, testis, brain, skin, fatty tissue, histocyte, macrophage, T cell, B cell). The HBR is total RNA isolated from the brains of 23 Caucasians, male and female, of varying age but mostly 60-80 years old.
In addition, a spike-in control was used. Specifically we added an aliquot of the ERCC ExFold RNA Spike-In Control Mixes to each sample. The spike-in consists of 92 transcripts that are present in known concentrations across a wide abundance range (from very few copies to many copies). This range allows us to test the degree to which the RNA-seq assay (including all laboratory and analysis steps) accurately reflects the relative abundance of transcript species within a sample. There are two ‘mixes’ of these transcripts to allow an assessment of differential expression output between samples if you put one mix in each of your two comparisons. In our case, Mix1 was added to the UHR sample, and Mix2 was added to the HBR sample. We also have 3 complete experimental replicates for each sample. This allows us to assess the technical variability of our overall process of producing RNA-seq data in the lab.
From the ERCC product sheet:
The ERCC plasmid reference library produces well-characterized transcripts generated largely from random unique sequences. Sequence comparisons have been made to multiple databases available at the time of design including mouse, rat, human, Drosophila, bacteria, mosquito, and other nonhuman species. The control collection contains some sequences with homology to Bacillus subtilis.
For all libraries we prepared low-throughput (Set A) TruSeq Stranded Total RNA Sample Prep Kit libraries with Ribo-Zero Gold to remove both cytoplasmic and mitochondrial rRNA. Triplicate, indexed libraries were made starting with 100ng Agilent/Strategene Universal Human Reference total RNA and 100ng Ambion Human Brain Reference total RNA. The Universal Human Reference replicates received 2 ul of 1:1000 ERCC Mix 1. The Human Brain Reference replicates received 2 ul of 1:1000 ERCC Mix 2. The libraries were quantified with KAPA Library Quantification qPCR and adjusted to the appropriate concentration for sequencing. The triplicate, indexed libraries were then pooled prior to sequencing. Each pool of three replicate libraries were sequenced across 2 lanes of a HiSeq 2000 using paired-end sequence chemistry with 100 bp read lengths.
So to summarize we have:
- UHR + ERCC Spike-In Mix1, Replicate 1
- UHR + ERCC Spike-In Mix1, Replicate 2
- UHR + ERCC Spike-In Mix1, Replicate 3
- HBR + ERCC Spike-In Mix2, Replicate 1
- HBR + ERCC Spike-In Mix2, Replicate 2
- HBR + ERCC Spike-In Mix2, Replicate 3
Each data set has a corresponding pair of FASTQ files (read 1 and read 2 of paired end reads).
echo $RNA_DATA_DIR
mkdir -p $RNA_DATA_DIR
cd $RNA_DATA_DIR
wget https://genomedata.org/rnaseq-tutorial/HBR_UHR_ERCC_ds_5pc.tar
Unpack the test data using tar. You should see 6 sets of paired end fastq files. One for each of our sample replicates above. We have 6 pairs (12 files) because in fastq format, read 1 and read 2 of a each read pair (fragment) are stored in separate files.
tar -xvf HBR_UHR_ERCC_ds_5pc.tar
ls
Enter the data directory and view the first two read records of a file (in fastq format each read corresponds to 4 lines of data)
The reads are paired-end 101-mers generated on an Illumina HiSeq instrument. The test data has been pre-filtered for reads that appear to map to chromosome 22. Lets copy the raw input data to our tutorial working directory.
zcat UHR_Rep1_ERCC-Mix1_Build37-ErccTranscripts-chr22.read1.fastq.gz | head -n 8
Identify the following components of each read: read name, read sequence, and quality string
How many reads are there in the first library? Decompress file on the fly with ‘zcat’, pipe into ‘grep’, search for the read name prefix and pipe into ‘wc’ to do a word count (‘-l’ gives lines)
zcat UHR_Rep1_ERCC-Mix1_Build37-ErccTranscripts-chr22.read1.fastq.gz | grep -P "^\@HWI" | wc -l
Determining the strandedness of RNA-seq data (Optional)
In order to determine strandedness, we will be using check_strandedness(docker image). In order use this tool, there are a few steps we need to get our inputs ready, specifically creating a fasta of our GTF file.
To create a transcripts FASTA file from our reference transcriptome in GTF format we need to get our transcript information into BED12 format. To get to that format we will convert GTF -> GenePred -> BED12. The GenePred format is just an intermediate that we create because we did not find a tool that would convert straight from GTF to BED12.
Once we have our transcriptome information (the position of exons for each transcript on each chromosome) we can use bedtools getfasta to splice the exon sequences from the chromosome sequence together into a full length transcript sequence.
cd $RNA_HOME/refs/
# Convert our reference transcriptome GTF to genePred format
gtfToGenePred chr22_with_ERCC92.gtf chr22_with_ERCC92.genePred
# Convert the genPred format to bed12 format
genePredToBed chr22_with_ERCC92.genePred chr22_with_ERCC92.bed12
# Use bedtools to create fasta from GTF
bedtools getfasta -fi chr22_with_ERCC92.fa -bed chr22_with_ERCC92.bed12 -s -split -name -fo chr22_ERCC92_transcripts.fa
Use less to view the file chr22_ERCC92_transcripts.fa. Note that this file has messy transcript names. Use the following hairball perl one-liner to tidy up the header line for each fasta sequence.
cd $RNA_HOME/refs
cat chr22_ERCC92_transcripts.fa | perl -ne 'if($_ =~/^\>\S+\:\:(ERCC\-\d+)\:.*/){print ">$1\n"}elsif ($_ =~/^\>(\S+)\:\:.*/){print ">$1\n"}else{print $_}' > chr22_ERCC92_transcripts.clean.fa
View the resulting ‘clean’ file using less chr22_ERCC92_transcripts.clean.fa (use ‘q’ to exit). View the end of this file using tail chr22_ERCC92_transcripts.clean.fa. Note that we have one fasta record for each Ensembl transcript on chromosome 22 and we have an additional fasta record for each ERCC spike-in sequence.
We also need to reformat our GTF file slightly. Rows that correspond to genes are missing the “transcript_id” field. We are going to add in this field but leave it blank for these rows using the following command.
cd $RNA_HOME/refs
awk '{ if ($0 ~ "transcript_id") print $0; else print $0" transcript_id \"\";"; }' chr22_with_ERCC92.gtf > chr22_with_ERCC92_tidy.gtf
Now that we have created our input files, we can now run the check_strandedness tool on some of our instrument data. Note: we are using a docker image for this tool.
docker run -v /home/ubuntu/workspace/rnaseq:/docker_workspace mgibio/checkstrandedness:latest check_strandedness --gtf /docker_workspace/refs/chr22_with_ERCC92_tidy.gtf --transcripts /docker_workspace/refs/chr22_ERCC92_transcripts.clean.fa --reads_1 /docker_workspace/data/HBR_Rep1_ERCC-Mix2_Build37-ErccTranscripts-chr22.read1.fastq.gz --reads_2 /docker_workspace/data/HBR_Rep1_ERCC-Mix2_Build37-ErccTranscripts-chr22.read2.fastq.gz
docker run is how you initialize a docker container to run a command
-v is the parameter used to mount your workspace so that the docker container can see the files that you’re working with. In the example above, /home/ubuntu/workspace/rnaseq from the EC2 instance has been mounted as /docker_workspace within the docker container.
mgibio/checkstrandedness is the docker container name. The :latest refers to the specific tag and release of the docker container.
The output of this command should look like so:
Fraction of reads failed to determine: 0.1123
Fraction of reads explained by "1++,1--,2+-,2-+": 0.0155
Fraction of reads explained by "1+-,1-+,2++,2--": 0.8722
Over 75% of reads explained by "1+-,1-+,2++,2--"
Data is likely RF/fr-firststrand
The checkstrandedness test asks a single question: in each read pair, which mate aligns in the same orientation as the gene it overlaps? The tool encodes every read as read# + aligned strand + gene strand, then sorts reads into two patterns. For the ‘reverse’ pattern 1+-,1-+,2++,2--: read 2 always agrees with the gene’s strand (2++, 2–) while read 1 always disagrees (1+-, 1-+). For the ‘forward’ pattern 1++,1--,2+-,2-+ is the exact mirror, with read 1 always agreeing and read 2 always disagreeing. The four codes within each pattern aren’t independent observations; they’re that one orientation rule applied to genes on the + strand and the - strand, which is why all four always appear together. In a dUTP/fr-firststrand prep, first-strand cDNA is synthesized antisense to the mRNA, the dUTP-marked second strand is degraded before amplification, and the adapters are arranged so read 1 sequences the surviving antisense strand while read 2 reads its sense complement. So read 2 matches the transcript and read 1 opposes it. The two names describe the same molecule from different angles: “first-strand” refers to the cDNA strand that is retained, and “reverse” refers to read 1 pointing opposite the transcript. In this case, 87.22% of reads fall in the reverse pattern, only 1.55% in the forward pattern, and 11.23% are undetermined (overlapping genes, multimappers, and ambiguous orientation, all expected and harmless). This strong bias toward 1+-,1-+,2++,2-- suggests an unambiguous RF / fr-firststrand call. A forward-stranded library would have put the bulk in the other bin. And an unstranded library would have split close to 50:50.
Using this table, we can see if this result is what we expect. Note that since the UHR and HBR data were generated with the TruSeq Stranded Kit, as mentioned above, the correct strand setting for kallisto is --rf-stranded, which is what check_strandedness confirms. Similarly when we run HISAT we will use --rna-strandness RF, when we run StringTie we will use --rf, and when we run htseq-count we will use --stranded reverse.
PRACTICAL EXERCISE 3
Assignment: Download an additional dataset and unpack it. This data will be used in future practical exercises.
- Hint: Do this in a separate working directory called ‘practice’ and create sub-directories for organization (data, alignments, etc).
- In this exercise you will download an archive of publicly available read data from here: https://genomedata.org/rnaseq-tutorial/practical.tar
The practice dataset includes 3 replicates of data from the HCC1395 breast cancer cell line and 3 replicates of data from HCC1395BL matched lymphoblastoid line. So, this will be a tumor vs normal (cell line) comparison. The reads are paired-end 151-mers generated on an Illumina HiSeq instrument from TruSeq Stranded Total RNA Libraries. The test data has been pre-filtered for reads that appear to map to chromosome 22.
Questions
- How many data files were contained in the ‘practical.tar’ archive? What commonly used sequence data file format are they?
- In the first read of the hcc1395, normal, replicate 1, read 1 file, what was the physical location of the read on the flow cell (i.e. lane, tile, x, y)?
- In the first read of this same file, how many ‘T’ bases are there?
Solution: When you are ready you can check your approach against the Solutions.
NOTE: The complete RAW HCC1395 data sets can be found here: https://genomedata.org/pmbio-workshop/fastqs/all/
If you use this data, please cite our paper: Citation