Posts
My thoughts and ideas
Welcome to the blog
My thoughts and ideas
Introduction to bioinformatics for RNA sequence analysis
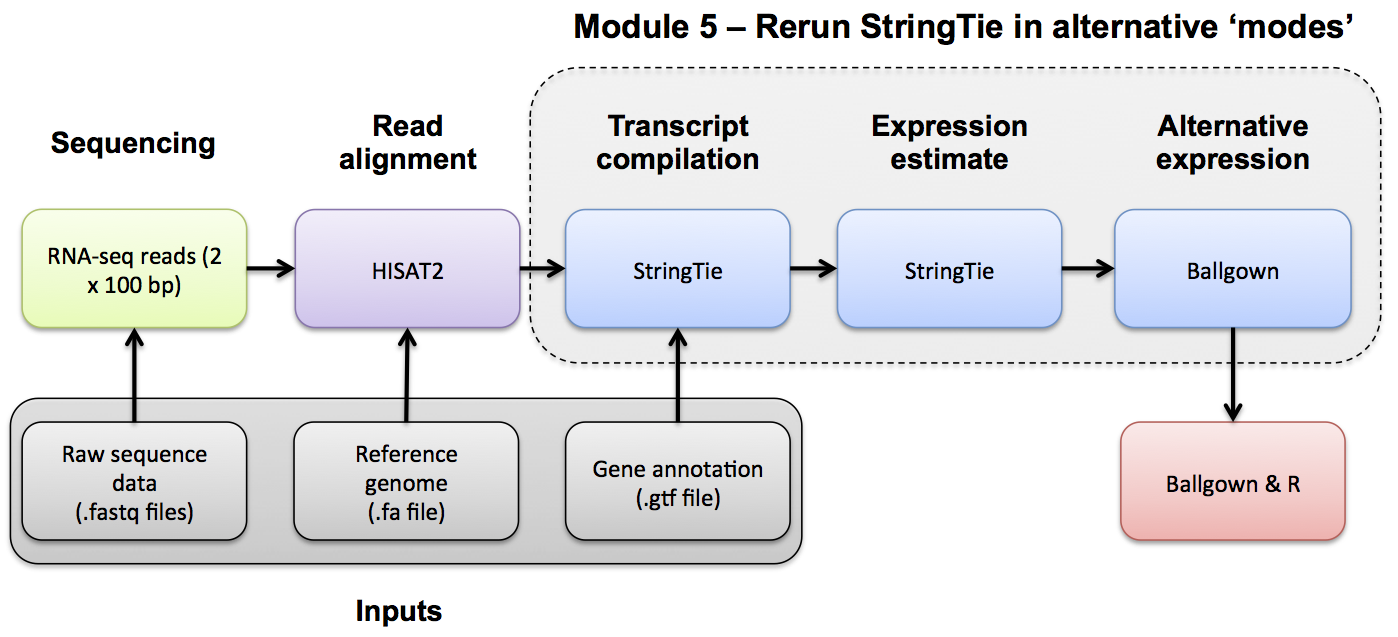
Note, to discover novel transcripts with Stringtie using the alignments we generated in the previous modules we will now run Stringtie in de novo mode. To use de novo mode do NOT specify either of the -G OR -e options.
Extra options specified below
–rf tells StringTie that our data is stranded and to use the correct strand specific mode (i.e. assume a stranded library fr-firststrand).-p 4 tells Stringtie to use eight CPUs-l name prefix for output transcripts (default: STRG)-o output path/file name for the assembled transcripts GTF (default: stdout)cd $RNA_HOME/
mkdir -p expression/stringtie/de_novo/
cd expression/stringtie/de_novo/
stringtie --rf -p 4 -l HBR_Rep1 -o HBR_Rep1/transcripts.gtf $RNA_ALIGN_DIR/HBR_Rep1.bam
stringtie --rf -p 4 -l HBR_Rep2 -o HBR_Rep2/transcripts.gtf $RNA_ALIGN_DIR/HBR_Rep2.bam
stringtie --rf -p 4 -l HBR_Rep3 -o HBR_Rep3/transcripts.gtf $RNA_ALIGN_DIR/HBR_Rep3.bam
stringtie --rf -p 4 -l UHR_Rep1 -o UHR_Rep1/transcripts.gtf $RNA_ALIGN_DIR/UHR_Rep1.bam
stringtie --rf -p 4 -l UHR_Rep2 -o UHR_Rep2/transcripts.gtf $RNA_ALIGN_DIR/UHR_Rep2.bam
stringtie --rf -p 4 -l UHR_Rep3 -o UHR_Rep3/transcripts.gtf $RNA_ALIGN_DIR/UHR_Rep3.bam