Posts
My thoughts and ideas
Welcome to the blog
My thoughts and ideas
Introduction to bioinformatics for RNA sequence analysis
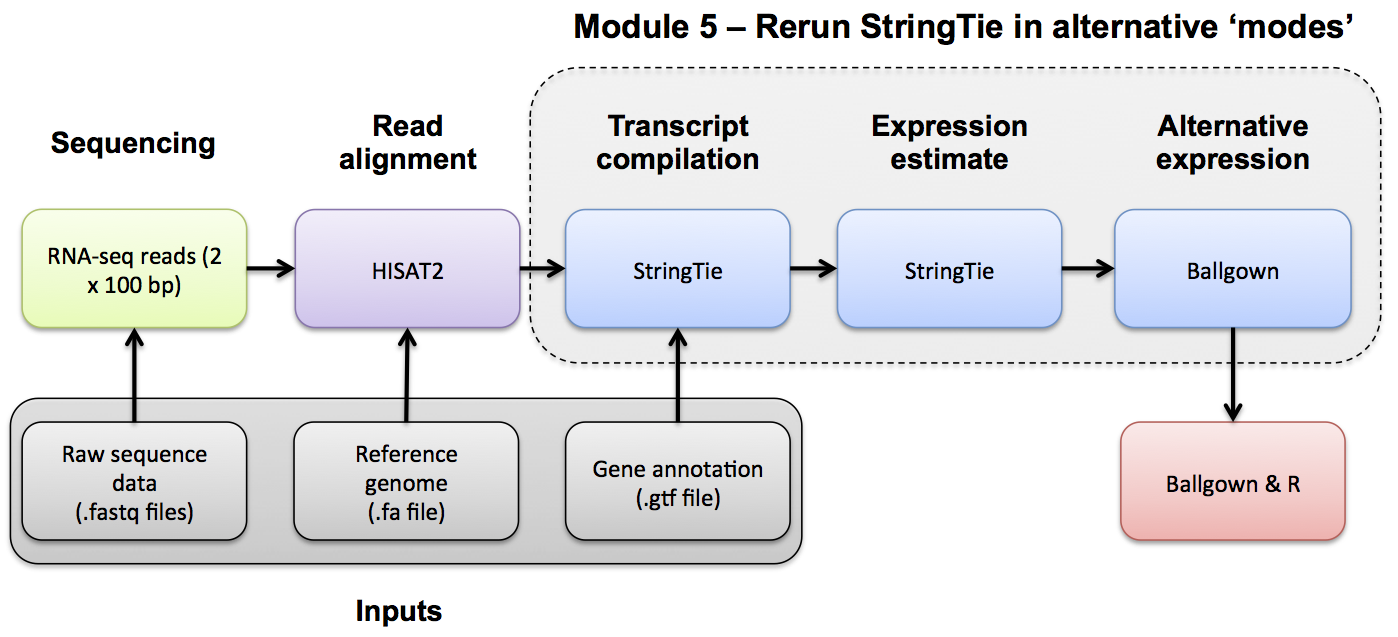
Use Ballgown and Stringtie to compare the UHR and HBR conditions against reference guided and de novo transcript assemblies.
Refer to the Stringtie manual for a more detailed explanation: https://ccb.jhu.edu/software/stringtie/index.shtml?t=manual
The Ballgown github page also has documentation for getting started with ballgown: https://github.com/alyssafrazee/ballgown
Re-run Stringtie using the reference guided merged GTF, and output tables for Ballgown. Store the results in a new directory so that we can still examine the results generated without the merged GTF.
cd $RNA_HOME/expression/stringtie/
mkdir ref_guided_merged
cd ref_guided_merged
stringtie --rf -p 4 -G ../ref_guided/stringtie_merged.gtf -e -B -o HBR_Rep1/transcripts.gtf $RNA_ALIGN_DIR/HBR_Rep1.bam
stringtie --rf -p 4 -G ../ref_guided/stringtie_merged.gtf -e -B -o HBR_Rep2/transcripts.gtf $RNA_ALIGN_DIR/HBR_Rep2.bam
stringtie --rf -p 4 -G ../ref_guided/stringtie_merged.gtf -e -B -o HBR_Rep3/transcripts.gtf $RNA_ALIGN_DIR/HBR_Rep3.bam
stringtie --rf -p 4 -G ../ref_guided/stringtie_merged.gtf -e -B -o UHR_Rep1/transcripts.gtf $RNA_ALIGN_DIR/UHR_Rep1.bam
stringtie --rf -p 4 -G ../ref_guided/stringtie_merged.gtf -e -B -o UHR_Rep2/transcripts.gtf $RNA_ALIGN_DIR/UHR_Rep2.bam
stringtie --rf -p 4 -G ../ref_guided/stringtie_merged.gtf -e -B -o UHR_Rep3/transcripts.gtf $RNA_ALIGN_DIR/UHR_Rep3.bam
Run Ballgown using the reference guided, merged transcripts
mkdir -p $RNA_HOME/de/ballgown/ref_guided_merged/
cd $RNA_HOME/de/ballgown/ref_guided_merged/
printf "\"ids\",\"type\",\"path\"\n\"UHR_Rep1\",\"UHR\",\"$RNA_HOME/expression/stringtie/ref_guided_merged/UHR_Rep1\"\n\"UHR_Rep2\",\"UHR\",\"$RNA_HOME/expression/stringtie/ref_guided_merged/UHR_Rep2\"\n\"UHR_Rep3\",\"UHR\",\"$RNA_HOME/expression/stringtie/ref_guided_merged/UHR_Rep3\"\n\"HBR_Rep1\",\"HBR\",\"$RNA_HOME/expression/stringtie/ref_guided_merged/HBR_Rep1\"\n\"HBR_Rep2\",\"HBR\",\"$RNA_HOME/expression/stringtie/ref_guided_merged/HBR_Rep2\"\n\"HBR_Rep3\",\"HBR\",\"$RNA_HOME/expression/stringtie/ref_guided_merged/HBR_Rep3\"\n" > UHR_vs_HBR.csv
Please see Differential Expression for details on running ballgown to determine a DE gene/transcript list.
Re-run Stringtie using the de novo merged GTF, and output tables for Ballgown. Store the results in a new directory so that we can still examine the results generated without the merged GTF.
cd $RNA_HOME/expression/stringtie/de_novo
cd $RNA_HOME/expression/stringtie/
mkdir de_novo_merged
cd de_novo_merged
stringtie --rf -p 4 -G ../de_novo/stringtie_merged.gtf -e -B -o HBR_Rep1/transcripts.gtf $RNA_ALIGN_DIR/HBR_Rep1.bam
stringtie --rf -p 4 -G ../de_novo/stringtie_merged.gtf -e -B -o HBR_Rep2/transcripts.gtf $RNA_ALIGN_DIR/HBR_Rep2.bam
stringtie --rf -p 4 -G ../de_novo/stringtie_merged.gtf -e -B -o HBR_Rep3/transcripts.gtf $RNA_ALIGN_DIR/HBR_Rep3.bam
stringtie --rf -p 4 -G ../de_novo/stringtie_merged.gtf -e -B -o UHR_Rep1/transcripts.gtf $RNA_ALIGN_DIR/UHR_Rep1.bam
stringtie --rf -p 4 -G ../de_novo/stringtie_merged.gtf -e -B -o UHR_Rep2/transcripts.gtf $RNA_ALIGN_DIR/UHR_Rep2.bam
stringtie --rf -p 4 -G ../de_novo/stringtie_merged.gtf -e -B -o UHR_Rep3/transcripts.gtf $RNA_ALIGN_DIR/UHR_Rep3.bam
Run Ballgown using the de novo, merged transcripts
mkdir -p $RNA_HOME/de/ballgown/de_novo_merged/
cd $RNA_HOME/de/ballgown/de_novo_merged/
printf "\"ids\",\"type\",\"path\"\n\"UHR_Rep1\",\"UHR\",\"$RNA_HOME/expression/stringtie/de_novo_merged/UHR_Rep1\"\n\"UHR_Rep2\",\"UHR\",\"$RNA_HOME/expression/stringtie/de_novo_merged/UHR_Rep2\"\n\"UHR_Rep3\",\"UHR\",\"$RNA_HOME/expression/stringtie/de_novo_merged/UHR_Rep3\"\n\"HBR_Rep1\",\"HBR\",\"$RNA_HOME/expression/stringtie/de_novo_merged/HBR_Rep1\"\n\"HBR_Rep2\",\"HBR\",\"$RNA_HOME/expression/stringtie/de_novo_merged/HBR_Rep2\"\n\"HBR_Rep3\",\"HBR\",\"$RNA_HOME/expression/stringtie/de_novo_merged/HBR_Rep3\"\n" > UHR_vs_HBR.csv
Please see Differential Expression for details on running ballgown to determine a DE gene/transcript list.